SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form
REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Or
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
Or
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Or
SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF |
Date of event requiring this shell company report________________________
For the transition period from__________ to ___________
Commission File No.
(Exact name of Registrant as specified in its charter)
Not Applicable
(Translation of Registrant’s name into English)
(Jurisdiction of incorporation or organization)
Telephone +44 020 7788 7414
(Address of principal executive offices)
Telephone
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
|
|
Securities registered or to be registered pursuant to Section 12(g) of the Act:
None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act:
None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report (March 31, 2024):
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. Yes ☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such a shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer |
☐ |
Accelerated filer |
☐ |
☒ |
|
|
|
|
|
Emerging growth company |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act
†The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
U.S. GAAP ☐ |
by the International Accounting Standards Board ☒ |
Other ☐ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow. Item 17 ☐ Item 18 ☐
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No
TABLE OF CONTENTS
1
69 |
2
FORWARD LOOKING STATEMENTS
This Annual Report on Form 20-F contains forward-looking statements, about our expectations, beliefs or intentions regarding, among other things, our product development efforts, business, financial condition, results of operations, strategies or prospects. In addition, from time to time, we or our representatives have made or may make forward-looking statements, orally or in writing. Forward-looking statements can be identified by the use of forward-looking words such as “believe,” “expect,” “intend,” “plan,” “may,” “should” or “anticipate” or their negatives or other variations of these words or other comparable words or by the fact that these statements do not relate strictly to historical or current matters. These forward-looking statements may be included in, but are not limited to, various filings made by us with the U.S. Securities and Exchange Commission, or the SEC, press releases or oral statements made by or with the approval of one of our authorized executive officers. Forward-looking statements relate to anticipated or expected events, activities, trends or results as of the date they are made. Because forward-looking statements relate to matters that have not yet occurred, these statements are inherently subject to risks and uncertainties that could cause our actual results to differ materially from any future results expressed or implied by the forward-looking statements. Many factors could cause our actual activities or results to differ materially from the activities and results anticipated in forward-looking statements, including, but not limited to, the factors summarized below.
This Annual Report on Form 20-F identifies important factors which could cause our actual results to differ materially from those indicated by the forward-looking statements, particularly those set forth under the heading “Risk Factors.” The risk factors included in this Annual Report on Form 20-F are not necessarily all the important factors that could cause actual results to differ materially from those expressed in any of our forward-looking statements. Given these uncertainties, readers are cautioned not to place undue reliance on such forward-looking statements. Factors that could cause our actual results to differ materially from those expressed or implied in such forward-looking statements include, but are not limited to:
All forward-looking statements attributable to us or persons acting on our behalf speak only as of the date of this Annual Report on Form 20-F and are expressly qualified in their entirety by the cautionary statements included in this Annual Report on Form 20-F. We undertake no obligations to update or revise forward-looking statements to reflect events or circumstances that arise after the date made or to reflect the occurrence of unanticipated events. In evaluating forward-looking statements, you should consider these risks and uncertainties.
3
CERTAIN DEFINITIONS
Unless otherwise indicated and except where the context otherwise requires, references in this Annual Report on Form 20-F to:
|
● |
“Exchange Act” refers to the United States Securities Exchange Act of 1934, as amended; |
|
● |
“FDA” refers to the United States Food and Drug Administration; |
|
● |
“GBP" refers to the British Pound; |
|
● |
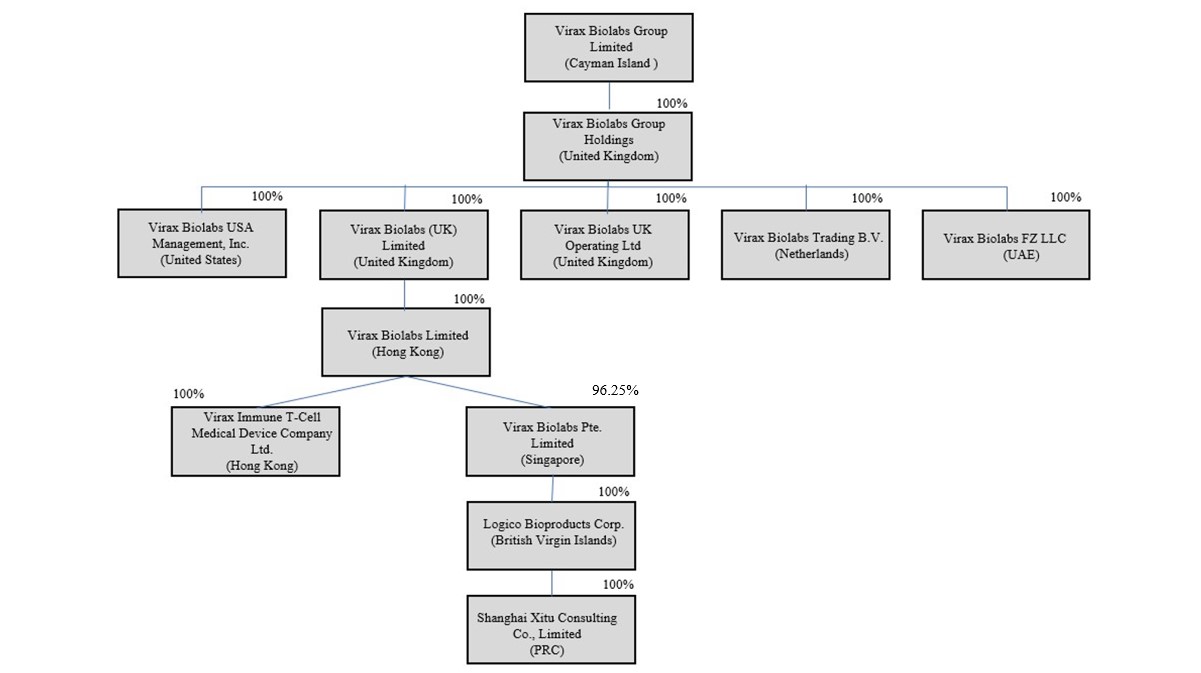
“HKco” refers to Virax Biolabs Limited, a wholly owned Hong Kong subsidiary of the Company, serving as a holding company; |
|
● |
“IVD” refers to in-vitro diagnostics; |
|
● |
“Logico BVI” refers to Logico Bioproducts Corp., a wholly-owned subsidiary of SingaporeCo incorporated in the British Virgin Islands; |
|
● |
“ordinary shares” refers to our ordinary shares, each of $0.001 par value; |
|
● |
“RMB” refers to the Renminbi; |
|
● |
“SEC” refers to the United States Securities and Exchange Commission; |
|
● |
“Shanghai Xitu” refers to Shanghai Xitu Consulting Co., Limited, a wholly-owned subsidiary of Logico BVI and a wholly foreign owned enterprise incorporated in China; |
|
● |
“SingaporeCo” refers to Virax Biolabs Pte. Limited, an operating subsidiary incorporated in Singapore; |
|
● |
“SGD” refers to the Singapore Dollar; |
|
● |
“Securities Act” refers to the Securities Act of 1933, as amended; |
|
● |
“Virax Biolabs,” the “Company,” “we,” “us” and “our” refer to Virax Biolabs Group Limited and our wholly owned subsidiaries; |
|
● |
“ViraxImmune T cell” refers to ViraxImmune T cell Medical Device Company Limited, a wholly-owned subsidiary of HKco; and |
|
● |
“$,” “USD,” “US$” and “U.S. dollar” refers to the United States dollar. |
4
PART I
Item 1. Identity of Directors, Senior Management and Advisers
Not applicable.
Item 2. Offer Statistics and Expected Timetable
Not applicable.
Item 3. Key Information
A. [Reserved]
B. Capitalization and indebtedness.
Not applicable.
C. Reasons for the offer and use of proceeds.
Not applicable.
D. Risk factors.
Risks Related to Our Business and Industry
We have limited operating history, have incurred operating losses for the years ended March 31, 2024 and 2023 and expect to incur significant losses for the foreseeable future. We may not generate sufficient revenue or become profitable or, if we achieve profitability, we may not be able to sustain it.
Biotechnology product development is a highly speculative undertaking and involves a substantial degree of risk. We are a clinical-stage biotechnology company with a limited operating history upon which you can evaluate our business and prospects. We commenced operations in 2013, and to date, we have focused primarily on organizing and staffing our company, business planning, raising capital, performing research and development activities, primarily the development of the ViraxImmune product and a mobile application, establishing our intellectual property portfolio, and conducting clinical trials.
We have incurred operating losses since inception. If our products are not successfully commercialized, namely, ViraxImmune, we may not generate further revenue. Our net losses were $6,739,120 and $5,457,763 for the years ended March 31, 2024 and 2023, respectively. As of March 31, 2024, we had an accumulated deficit of $18,527,997. Substantially all of our losses have resulted from expenses incurred in connection with our research and development programs and from general and administrative costs associated with our operations. ViraxImmune products will require additional development time and resources before we would begin generating revenue from product sales. We expect to continue to incur losses for the foreseeable future, and we anticipate these losses will increase substantially as we conduct our ongoing research and development of our ViraxImmune products and seek to obtain product certification approvals in the territories we have identified, as well as hire additional personnel, obtain and protect our intellectual property and incur additional costs for commercialization or to expand our pipeline of product candidates.
To become and remain profitable, we must succeed in developing and eventually commercializing products that generate sufficient revenue. This will require us to be successful in a range of challenging activities, including completing preclinical studies and clinical trials of our product candidates, obtaining product certification approvals in the territories we have identified and manufacturing, marketing and selling any products for which we obtained product certification approvals. We may never succeed in these activities and, even if we do, may never generate revenues that are sufficient enough to achieve profitability. In addition, we have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biotechnology industry. Because of the numerous risks and uncertainties associated with biotechnology product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would depress the value of our company and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our product candidates or even continue our operations. A decline in the value of our company could also cause you to lose all or part of your investment.
5
We expect to make significant investments in our continued research and development of new products and services, which may not be successful.
We are seeking to build upon our existing research and development to develop a comprehensive set of T cell diagnostics and immune profiling solutions. Our strategic focus is the development and commercialization of immune profiling IVDs in indications associated with chronic inflammation and T cell exhaustion, such as post viral syndromes and other infectious diseases, chronic inflammation and immune-oncology.
Developing new products and services is a speculative and risky endeavor. Products or services that initially show promise may fail to achieve the desired results or may not achieve acceptable levels of analytical accuracy or clinical utility. We may need to alter our products in development and repeat clinical studies before we identify a potentially successful product or service. Product development is expensive, may take years to complete and can have uncertain outcomes. Failure can occur at any stage of the development. If, after development, a product or service appears successful, we may, depending on the nature of the product or service, still need to obtain regulatory clearances, authorizations or approvals before we can market it. The regulatory clearance, authorization or approval pathways are likely to involve significant time, as well as additional research, development and clinical study expenditures. The regulatory authorities may not clear, authorize or approve any future product or service we develop. Even if we develop a product or service that receives regulatory clearance, authorization or approval, we would need to commit substantial resources to commercialize, sell and market it before it could be profitable, and the product or service may never be commercially successful. Additionally, development of any product or service may be disrupted or made less viable by the development of competing products or services.
New potential products and services may fail at any stage of development or commercialization and if we determine that any of our current or future products or services are unlikely to succeed, we may abandon them without any return on our investment. If we are unsuccessful in developing additional products or services, our potential for growth may be impaired, and our business, financial condition and results of operations may be adversely affected.
If we are not successful in leveraging the ViraxImmune platform to discover, develop and commercialize additional products and services, our ability to expand our business and achieve our strategic objectives would be impaired.
A key element of our strategy is to leverage our ViraxImmune platform to address a wide range of research, diagnostic and health needs within indications associated with chronic inflammation and T cell exhaustion through our test kits. If we are unable to generate compelling evidence supporting our T cell test results, our platform may face a broader obstacle to using our diagnostics data for commercially viable products and services.
Identifying new products and services requires substantial technical, financial and human resources, whether or not any products or services are ultimately developed or commercialized. We may pursue what we believe is a promising opportunity to leverage our platform only to discover that certain of our risk or resource allocation decisions were incorrect or insufficient, or that individual products, services or our science in general has technology or biology risks that were previously unknown or underappreciated. Our strategy of pursuing the value of our diagnostics platform over a long time horizon and developing relevant technological products with synergy may not be effective. In the event material decisions in any of these areas turn out to be incorrect or sub-optimal, we may experience a material adverse impact on our business and ability to fund our operations and we may never realize what we believe is the potential of our in-vitro diagnostics platform.
Our efforts to develop a T cell In-Vitro Diagnostic Test may not be successful, and it may not yield the insights we expect at all or on a timetable that allows us to develop or commercialize any new diagnostic products.
We are currently developing a comprehensive set of T cell diagnostics and immune profiling solutions. Our strategic focus is the development and commercialization of immune profiling IVDs in indications associated with chronic inflammation and T cell exhaustion, such as post viral syndromes and other infectious diseases, chronic inflammation and immune-oncology.
ViraxImmune may not yield clinically actionable insights on a timetable that is commercially viable, or at all. ViraxImmune will initially be focusing in diseases associated with post viral syndromes including but not limited to SARS-CoV-2, Human Papillomavirus (better known as HPV), Malaria, Hepatitis B, and Herpes (better known as HSV-1). If our efforts do not accelerate the pace at which we can validate our diagnostic method, the timetable for our business model may not be commercially viable. Even if we can accelerate this timeline, our products and services derived from our novel technologies may have product or service level errors. If we are unable to make meaningful progress in our technology and successfully use it to develop and commercialize new diagnostic products or services, our business and results of operations will suffer.
If we are not successful in obtaining regulatory approvals for our ViraxImmune products, we may not be able to commercialize our products in the expected timeframe or at all, and our ability to expand our business and achieve our strategic objectives would be impaired.
6
Currently, we are developing a comprehensive set of T cell diagnostics and immune profiling solutions. Our strategic focus is the development and commercialization of immune profiling IVDs in indications associated with chronic inflammation and T cell exhaustion, such as post viral syndromes and other infectious diseases, chronic inflammation and immune-oncology. For example, in the United States, the FDA regulates the sale or distribution of medical devices, including but not limited to, IVD test kits. IVD products are subject to regulation by the FDA as medical devices to the extent that they are intended for use in the diagnosis, treatment, mitigation or prevention of disease or other conditions. They are subject to premarket review and post market controls which will differ depending on how the FDA classifies a specific IVD.
The information that must be submitted to the FDA in order to obtain clearance or approval to market a new medical device varies depending on how the medical device is classified by the FDA. Medical devices are classified into one of three classes depending on the controls deemed by the FDA to be necessary to reasonably ensure their safety and effectiveness. Class I devices are subject to general controls, including labeling requirements, and adherence to the FDA’s quality system regulations, or QSRs, which are device-specific current good manufacturing practices. Class II devices are subject to premarket notification, QSRs, general controls and sometimes special controls, including performance standards and post-market surveillance. Class III devices are subject to most of the previously identified requirements as well as to pre-market approval. Class I devices are exempt from premarket review; most Class II devices require 510(k) clearance, and all Class III devices must receive premarket approval before they can be sold in the United States.
A 510(k) premarket notification requires the sponsor to demonstrate that a medical device is substantially equivalent to another marketed device, termed a “predicate device,” that is legally marketed in the United States and for which a premarket approval was not required. A device is substantially equivalent to a predicate device if it has the same intended use and technological characteristics as the predicate; or has the same intended use but different technological characteristics, where the information submitted to the FDA does not raise new questions of safety and effectiveness and demonstrates that the device is at least as safe and effective as the legally marketed device.
A Premarket Approval process is more complex, costly and time consuming than the 510(k) process. A PMA must be supported by more detailed and comprehensive scientific evidence, including clinical data, to demonstrate the safety and efficacy of the medical device for its intended purpose. If the device is determined to present a “significant risk,” the sponsor may not begin a clinical trial until it submits an investigational device exemption (IDE) to the FDA and obtains approval to begin the trial.
Should we fail to obtain the necessary FDA or the relevant regulatory authority’s approval, for example, to demonstrate to the FDA or the relevant regulatory authority’s satisfaction that our T cell IVD/Immune response Test kits are safe and effective, we may not be able to commercialize our ViraxImmune product and/or platform in the expected timeframe or at all, and our ability to expand our business and achieve our strategic objectives would be impaired.
We will face significant challenges in successfully commercializing our products, particularly in new markets.
We intend to grow our own sales and marketing capabilities and promote our product candidates if and when regulatory approval has been obtained in the United Kingdom, European Union and North America, and to expand to other markets as well. In order to successfully commercialize our products in these new markets, we require appropriate infrastructure such as information technology, enterprise resource planning and forecasting. We may fail to launch our products effectively or to market our products effectively. Recruiting and training a sales force is expensive and costs of creating an independent sales and marketing organization and of marketing and promotion could be above what we anticipate. In addition, recruiting and training a sales force is time consuming and could delay any product launch. In the event that any such launch is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
If we enter into arrangements with third parties to perform sales and marketing services, our product revenues or the profitability of these product revenues to us could be lower than if we were to market and sell any products that we develop ourselves. Such collaborative arrangements may place the commercialization of our products outside of our control and would make us subject to a number of risks, including that we may not be able to control the amount or timing of resources that our collaborative partner devotes to our products or our collaborator’s willingness or ability to complete its obligations, and our obligations under our arrangements may be adversely affected by business combinations or significant changes in our collaborator’s business strategy. In addition, we may not be successful in entering into arrangements with third parties to sell and market our products or may be unable to do so on terms that are favorable to us. Acceptable third parties may fail to devote the necessary resources and attention to sell and market our products effectively.
If we do not establish sales and marketing capabilities in new markets successfully in our targeted expansion regions or countries, either on our own or in collaboration with third parties, we may not be successful in commercializing our products, which in turn would have a material adverse effect on our business, financial condition and results of operations.
Our business, financial condition and results of operations will depend on the market acceptance and increased demand for our products by Contract Research Organizations, hospitals, governments and public health departments, as well as physicians and
7
others in the medical community, and the growing proportion of the population who are interested in taking personal charge over their health and wellbeing.
Our future success depends on our products gaining sufficient market acceptance by hospitals, public health departments and consumer groups interested in their health and wellbeing. If our products do not achieve an adequate level of acceptance by such customer groups, we may not generate enough revenue to become profitable. For example, the degree of market acceptance of our T cell in-vitro diagnostics product will depend on a number of factors, including:
Our efforts to educate physicians and other members of the medical community on the benefits of our products may require significant resources and may never be successful. Such efforts to educate the marketplace may require more resources than are required by conventional technologies marketed by our competitors. In particular, continuing to gain market acceptance for our products in nascent markets could be challenging. In certain markets, including, for example Canada and United States, our potential for future growth is difficult to forecast. If we were to incorrectly forecast our ability to penetrate these markets, expenditures that we make may not result in the benefits that we expect, which could harm our results of operations. Additionally, if we lose any of our customers due to significant delays in our ability to obtain re-registration of our T cell IVD/Immune response Test in our initial target markets, our results of operations could be materially and adversely affected.
In the event that our products are the subject of guidelines, clinical studies or scientific publications that are unhelpful or damaging, or otherwise call into question the benefits of our products, we may have difficulty in convincing prospective customers to adopt our test. Moreover, the perception by the investment community or shareholders that recommendations, guidelines or studies will result in decreased use of our products could adversely affect the prevailing market price for our ordinary shares. Similar challenges apply to all of the products in our pipeline.
The success of some of our products partially depends on the continued demand for diagnostic products linked to SARS-CoV-2 and other major viral diseases.
Even if we achieve market acceptance, our success will partially depend on continued demand for diagnostic products for SARS-CoV-2 and other major viral diseases. SARS-CoV-2 screening policies could change such that tests are conducted less frequently or in fewer instances. For example, healthcare institutions facing increased cost control requirements could determine to reduce employee testing. In addition, various institutions or governing bodies may decide that the incidence of SARS-CoV-2 has dropped sufficiently in the future within their screening population so as to permit reduced testing. Changes to immigration policies and policies relating to resettlement of refugees, as well as other policy changes may substantially reduce testing in the markets we serve and could have a material and adverse effect on our business. In order to reduce our dependency on continued demand for diagnostic products in relation to SARS-CoV-2, we are developing our technology to focus on other major viral threats, however, we cannot be sure whether such developments can be successful. If we fail to develop our technology to easily adapt to new variants of coronavirus or potential new viral threats, it may materially adversely affect our financial condition and results of operations.
The success of our proprietary technology T cell testing requires us to proceed through clinical and validation studies successfully, which is not guaranteed.
In order for our proprietary technology T cell IVD/Immune response Test to be successful, we are required to sucessfully proceed through further clinical and validation studies, which is not guaranteed. Clinical testing or validation is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time and may adversely affect our operations and finances should there be a prolonged process of clinical and validation studies.
New market opportunities may not develop as quickly as we expect, limiting our ability to market and sell our products successfully.
We intend to take steps to continue to increase the presence of our products in markets both in the target markets and in the wider international market including the United Kingdom, EU, United States and Canada. We intend to grow our sales force globally and
8
establish distributor relationships to better access international markets. We believe these opportunities will take substantial time to develop or mature, however, and we cannot be certain that these market opportunities will develop as we expect. The future growth and success of our products in these markets depends on many factors beyond our control, including recognition and acceptance by the scientific community in that market and the prevalence and costs of competing methods of viral screening. If the markets for our products do not develop as we expect, our business may be adversely affected.
Our efforts to discover and develop products and services related to the ViraxImmune products may not be successful from either a platform extension or commercialization perspective.
We are in the process of developing a comprehensive set of T-cell diagnostics and immune profiling solutions. The platform's capabilities will initially focus on measuring chronic inflammation associated with T-cell exhaustion in areas such as Long COVID effects, Chimeric Antigen Receptor T-cell (CAR-T) therapies, Myloid, Encephalomyelitis (ME) and post viral syndromes. We are still in the process of conducting further tests and we have not submitted any clinical performance studies to any regulatory agency for approval. While we believe quantifying virus-specific T cells may provide important research and diagnostic advantages because T cells persist in the immune system later than antibodies, the data upon which such belief is based is limited and our analyses are preliminary. As we continue to collect and analyze additional data, we may find that our initial hypotheses are not applicable to some major viral diseases, or are not supported by a larger data set or further analysis. If our beliefs regarding the effectiveness of T cells in-vitro diagnostics tests are incorrect, that could have a material adverse effect on the market for T cells in-vitro diagnostics tests, our revenue expectations, reputation, financial condition, and our stock price, which would be adversely impacted.
Our efforts to further develop and commercialize T cell diagnostics tests involve a high degree of risk, and our efforts may fail for many reasons, including:
Additionally, there can be no assurances as to the commercial success of T cell in-vitro diagnostics tests for major viral disease. Our investments in the discovery and development of products and services related to major viral disease may not be accretive to our future financial results and if we determine that any product or service is unlikely to succeed, we may abandon them without any return on our investment.
We may be liable for improper collection, use or appropriation of personal information provided by our customers.
We collect certain personal data from our customers in target markets in connection with our business and operations, and we may expand our collection of data into areas including genetic data. Our collection of customer data is subject to various regulatory requirements relating to the security and privacy of data in various jurisdictions. Regulatory requirements regarding the protection of data are constantly evolving and can be subject to different interpretations or significant change, making the extent of our responsibilities in that regard uncertain.
In Europe, Directive 95/46/EC of the European Parliament and of the Council of October 24, 1995 on the protection of individuals with regard to the processing of personal data and on the free movement of such data, or the Directive, and Directive 2002/58/EC of the European Parliament and of the Council of July 12, 2002 concerning the processing of personal data and the protection of privacy in the electronic communications sector (as amended by Directive 2009/136/EC), or the e-Privacy-Directive, have required the European Union, or EU member states, to implement data protection laws to meet strict privacy requirements. Violations of these requirements can result in administrative measures, including fines, or criminal sanctions. The e-Privacy Directive will likely be replaced in time by a new e-Privacy Regulation which may impose additional obligations and risk for our business.
Beginning on May 25, 2018, the Directive was replaced by Regulation (EU) 2016/679 of the European Parliament and of the Council of April 27, 2016 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data, or the GDPR. The GDPR imposes a broad range of strict requirements on companies subject to the GDPR, such as us, including requirements relating to having legal bases for processing personal information relating to identifiable individuals and transferring such information outside the European Economic Area, or the EEA, including to the United States, providing details to those individuals regarding the processing of their personal information, keeping personal information secure, having data processing agreements with
9
third parties who process personal information, responding to individuals’ requests to exercise their rights in respect of their personal information, reporting security breaches involving personal data to the competent national data protection authority and affected individuals, appointing data protection officers, conducting data protection impact assessments, and record-keeping. The GDPR substantially increases the penalties to which we could be subject in the event of any non-compliance, including fines of up to 10,000,000 Euros or up to 2% of our total worldwide annual turnover for certain comparatively minor offenses, or up to 20,000,000 Euros or up to 4% of our total worldwide annual turnover for more serious offenses. We face uncertainty as to the exact interpretation of the requirements under the GDPR, and we may be unsuccessful in implementing all measures required by data protection authorities or courts in interpretation of the GDPR.
In particular, national laws of member states of the EU are in the process of being adapted to the requirements under the GDPR, thereby implementing national laws which may partially deviate from the GDPR and impose different obligations from country to country, so that we do not expect to operate in a uniform legal landscape in the EU. In the future, should we collect any genetic data in connection with our business and operations, our operations may also be subject to the GDPR, which specifically allows national laws to impose additional and more specific requirements or restrictions, and European laws have historically differed quite substantially in this field, leading to additional uncertainty.
We expect that we will continue to face uncertainty as to whether our efforts to comply with our obligations under European privacy laws will be sufficient. If we are investigated by a European data protection authority, we may face fines and other penalties. Any such investigation or charges by European data protection authorities could have a negative effect on our existing business and on our ability to attract and retain new clients or pharmaceutical partners. We may also experience hesitancy, reluctance, or refusal by European or multi-national clients or pharmaceutical partners to continue to use our products and solutions due to the potential risk exposure as a result of the current (and, in particular, future) data protection obligations imposed on them by certain data protection authorities in interpretation of current law, including the GDPR. Such clients or pharmaceutical partners may also view any alternative approaches to compliance as being too costly, too burdensome, too legally uncertain, or otherwise objectionable and therefore decide not to do business with us. Any of the foregoing could materially harm our business, prospects, financial condition and results of operations.
In Singapore, under the Personal Data Protection Act 2012 (the “PDPA”), we are required to, among others, notify individuals of the purposes for the collection, use or disclosure of their personal data prior to such collection, and to also disclose and obtain the consent of individuals during the collection, use or disclosure of their personal data.
A part of our operations is also carried out in China and a portion of the data and personal information we collected will need to be stored in China where relevant to ensure compliance with the Peoples Republic of China ("PRC") laws. We do not hold personal information of more than one million users and we believe that the Company’s initial public offering (“IPO”) of ordinary shares in July 2022 was not subject to PRC cybersecurity review. In addition, as of the date of this report, we have not received any notice of and are not currently subject to any proceedings initiated by the CAC or any other PRC regulatory authority. In addition, we may be subject to heightened regulatory scrutiny from PRC governmental authorities in the future. As there remains significant uncertainty in the interpretation and enforcement of the Data Security Law and the PRC Personal Information Protection Law (the "PIPL"), we cannot assure you that we will comply with such regulations in all respects. Any non-compliance with these laws and regulations may subject us to fines, orders to rectify or terminate any actions that are deemed illegal by regulatory authorities, other penalties, including but not limited to reputational damage or legal proceedings against us, which may affect our business, financial condition or results of operations.
The in-vitro diagnostics industry is subject to rapid change, which could make our diagnostics platform and related products and services that we develop obsolete.
Our industry is characterized by rapid changes, including technological and scientific breakthroughs, frequent new product and service introductions and enhancements and evolving industry standards, all of which could make our current and future products and services obsolete. Our future success will depend on our ability to keep pace with the evolving needs of our customers on a timely and cost-effective basis and to pursue new market opportunities that develop as a result of scientific and technological advances. In recent years, there have been numerous advances in technologies relating to the diagnosis and treatment of viral diseases. There have also been advances in technologies used to computationally analyze very large amounts of biologic information. If we do not update our products and services to reflect new scientific knowledge about diagnostics technology, software development, our products and services could become obsolete and any sales of our current products and services and any future products and services we develop based on our diagnostics platform could decline or fail to grow as expected.
Our business could suffer if we lose the services of, or are unable to attract and retain, key members of our senior management, key advisors or other personnel.
We are dependent upon the continued services of key members of our senior management, including James Foster, Nigel McCracken and Jason Davis. The loss of any one of these individuals, without adequate time to find a suitable replacement, could disrupt our operations or our strategic plans. Additionally, our future success will depend on, among other things, our ability to continue to hire and
10
retain the necessary qualified scientific, technical, sales, marketing and managerial personnel, for whom we compete with numerous other companies, academic institutions and organizations. The loss of members of our management team, key advisors or personnel, or our inability to attract or retain other qualified personnel or advisors, could have a material adverse effect on our business, results of operations and financial condition. Although all members of our senior management team have entered into agreements that restrict their ability to compete with us for a period of time after the end of their employment, we may be unable to enforce such restrictive covenants at all or for a sufficient duration of time to prevent members of our management team from competing against us.
We depend on our information technology systems and any failure of these systems could harm our business.
We depend on information technology and telecommunications systems, including third-party cloud computing infrastructure and operating systems, for significant elements of our operations, including our products research and development and e-commerce platform development.
We use complex software processes to manage and test samples and evaluate the resulting data. These are subject to initial design or ongoing modifications which may result in unanticipated issues that could cause variability in patient results, leading to service disruptions or errors, and resulting in liability.
We have installed, and expect to expand, a number of enterprise software systems that affect a broad range of business processes and functional areas, including systems handling human resources, financial controls and reporting, contract management, regulatory compliance and other infrastructure operations. In addition to these business systems, we have installed, and intend to extend, the capabilities of both our preventative and detective cybersecurity controls by augmenting the monitoring and alerting functions, the network design and the automatic countermeasure operations of our technical systems. These information technology and telecommunications systems will support a variety of functions, including laboratory operations, test validation, sample tracking, quality control, customer service support, billing and reimbursement, research and development activities, scientific and medical curation and general administrative activities. In addition, our third-party billing and collections provider depends upon technology and telecommunications systems provided by outside vendors.
Information technology and telecommunications systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts (such as ransomware) and natural disasters. Moreover, despite network security and back-up measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we have taken to prevent unanticipated problems that could affect our information technology and telecommunications systems, failures or significant downtime of these systems or those used by our partners or subcontractors could prevent us from conducting our diagnostic products development, completing the tests on our customer samples, preparing and providing reports to researchers, clinicians and our partners, billing and payments, handling enquiries, conducting research and development activities and managing the administrative aspects of our business. Any disruption or loss of information technology or telecommunications systems on which critical aspects of our operations depend could have an adverse effect on our business and our reputation, and we may be unable to regain or repair our reputation in the future.
We face risks related to natural disasters, health epidemics and other outbreaks which could significantly disrupt our operations.
In general, our business could be adversely affected by the effects of epidemics, including, but not limited to, SARS-CoV-2, avian influenza, severe acute respiratory syndrome (SARS), the influenza A virus, Ebola virus, severe weather conditions such as a snowstorm, flood or hazardous air pollution, or other outbreaks. In response to an epidemic, severe weather conditions, or other outbreaks, government and other organizations may adopt regulations and policies that could lead to severe disruption to our daily operations, including temporary closure of our offices and other facilities. These severe conditions may cause us and/or our partners to make internal adjustments, including but not limited to, temporarily closing down business, limiting business hours, and setting restrictions on travel and/or visits with clients and partners for a prolonged period of time. Various impacts arising from severe conditions may cause business disruption, resulting in material, adverse impact to our financial condition and results of operations.
Risks Related to Intellectual Property
If we are not able to adequately protect our proprietary intellectual property and information, and protect against third party claims that we are infringing on their intellectual property rights, our results of operations could be adversely affected.
The value of our business depends in part on our ability to protect our intellectual property and information, including our patents, copyrights, trademarks, trade secrets, and rights under agreements with third parties, in the United Kingdom and around the world, as well as our customers, employees, and customer data. Third parties may try to challenge our ownership of our intellectual property globally. In addition, intellectual property rights and protections in the United Kingdom may be insufficient to protect material intellectual property rights globally and the United Kingdom. Further, our business is subject to the risk of third parties counterfeiting our products or infringing on our intellectual property rights. The steps we have taken may not prevent unauthorized use of our intellectual property. We may need to resort to litigation to protect our intellectual property rights, which could result in substantial costs
11
and diversion of resources. If we fail to protect our proprietary intellectual property and information, including with respect to any successful challenge to our ownership of intellectual property or material infringements of our intellectual property, this failure could have a significant adverse effect on our business, financial condition, and results of operations.
If we are unable to adequately protect our intellectual property rights, or if we are accused of infringing on the intellectual property rights of others, our competitive position could be harmed or we could be required to incur significant expenses to enforce or defend our rights.
Our commercial success will depend in part on our success in obtaining and maintaining patents, copyrights, trademarks, trade secrets and other intellectual property rights in Europe and elsewhere and protecting our proprietary technology. If we do not adequately protect our intellectual property and proprietary technology, competitors may be able to use our technologies or the goodwill we have acquired in the marketplace and erode or negate any competitive advantage we may have, which could harm our business and ability to achieve profitability.
We cannot be certain that patents will be issued or granted with respect to applications that are currently pending. As a biotechnology company, our patent position is uncertain because it involves complex legal and factual considerations. The standards applied by the European Patent Office, the United States Patent and Trademark Office, or USPTO, and foreign patent offices in granting patents are not always applied uniformly or predictably. For example, there is no uniform worldwide policy regarding patentable subject matter or the scope of claims allowable in biotechnology patents. Consequently, patents may not issue from our pending patent applications. As such, we do not know the degree of future protection that we will have on our proprietary products and technology. The scope of patent protection that the European Patent Office and the USPTO will grant with respect to the antibodies in our antibodies product pipeline is uncertain. It is possible that the European Patent Office and the USPTO will not allow broad antibody claims that cover antibodies closely related to our product candidates as well as the specific antibody. As a result, upon receipt of European Medicines Agency or Food and Drug Administration approval, competitors may be free to market antibodies almost identical to ours, including biosimilar antibodies, thereby decreasing our market potential. However, a competitor cannot submit to the European Medicines Agency or Food and Drug Administration an application for a biosimilar product based on one of our products until four years following the date of approval of our “reference product,” and the European Medicines Agency or Food and Drug Administration may not approve such a biosimilar product until 12 years from the date on which the reference product was approved.
We cannot provide any assurances that any of our patents have, or that any of our pending patent applications that mature into issued patents will include, claims with a scope sufficient to protect our products, any additional features we develop for our products or any new products. Other parties may have developed technologies that may be related or competitive to our system, may have filed or may file patent applications and may have received or may receive patents that overlap or conflict with our patent applications, either by claiming the same methods or devices or by claiming subject matter that could dominate our patent position. Our patent position may involve complex legal and factual questions, and, therefore, the scope, validity and enforceability of any patent claims that we may obtain cannot be predicted with certainty. Patents, if issued, may be challenged, deemed unenforceable, invalidated or circumvented. Proceedings challenging our patents could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such proceedings may be costly. Thus, any patents that we may own may not provide any protection against competitors. Furthermore, an adverse decision in an interference proceeding can result in a third party receiving the patent right sought by us, which in turn could affect our ability to commercialize our products.
Though an issued patent is presumed valid and enforceable, its issuance is not conclusive as to its validity or its enforceability and it may not provide us with adequate proprietary protection or competitive advantages against competitors with similar products. Competitors could purchase our products and attempt to replicate some or all of the competitive advantages we derive from our development efforts, willfully infringe our intellectual property rights, design around our patents, or develop and obtain patent protection for more effective technologies, designs or methods. We may be unable to prevent the unauthorized disclosure or use of our technical knowledge or trade secrets by consultants, suppliers, vendors, former employees and current employees.
Our ability to enforce our patent rights depends on our ability to detect infringement. It may be difficult to detect infringers who do not advertise the components that are used in their products. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded if we were to prevail may not be commercially meaningful.
In addition, proceedings to enforce or defend our patents could put our patents at risk of being invalidated, held unenforceable or interpreted narrowly. Such proceedings could also provoke third parties to assert claims against us, including that some or all of the claims in one or more of our patents are invalid or otherwise unenforceable. If any of our patents covering our products are invalidated or found unenforceable, or if a court found that valid, enforceable patents held by third parties covered one or more of our products, our competitive position could be harmed or we could be required to incur significant expenses to enforce or defend our rights.
The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
12
We rely, in part, upon unpatented trade secrets, unpatented know-how and continuing technological innovation to develop and maintain our competitive position. Further, our trade secrets could otherwise become known or be independently discovered by our competitors.
We intend to apply for patents in the United States, subject to approval from the relevant regulatory bodies. If we do not obtain protection under the Hatch-Waxman Amendments and similar non-U.S. legislation for extending the term of patents covering each of our product candidates, our business may be materially harmed.
We consider the United States as a target market with significant potential. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if future patents covering our product candidates, their manufacture, or use are obtained, once the patent life has expired, we may be open to competition from competitive diagnostics, including biosimilar diagnostics. Given the amount of time required for the development, testing and regulatory review of new product candidates, future patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our future owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
Depending upon the timing, duration and conditions of future FDA marketing approval of our product candidates, one or more of our future U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Act and similar legislation in the European Union. The Hatch-Waxman Act permits a patent term extension of up to five years for a patent covering an approved product as compensation for effective patent term lost during product development and the FDA regulatory review process. The patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and only one patent applicable to an approved drug may be extended. However, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Moreover, the length of the extension could be less than we request. If we are unable to obtain future patent term extension or the term of any such extension is less than we request, the period during which we can enforce our future patent rights for that product will be shortened and our competitors may obtain approval to market competing products sooner than we expect. As a result, our revenue from applicable products could be reduced, possibly materially.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage and changes in patent laws or patent jurisprudence could diminish the value of patents in general, thereby impairing our ability to protect our products.
The America Invents Act, or the AIA, has been enacted in the United States, resulting in significant changes to the U.S. patent system. An important change introduced by the AIA is that, as of March 16, 2013, the United States transitioned to a “first-to-file” system for deciding which party should be granted a patent when two or more patent applications are filed by different parties claiming the same invention. A third party that files a patent application in the USPTO after that date but before us could therefore be awarded a patent covering an invention of ours even if we had made the invention before it was made by the third party. This will require us to be cognizant going forward of the time from invention to filing of a patent application, but circumstances could prevent us from promptly filing patent applications on our inventions.
Among some of the other changes introduced by the AIA are changes that limit where a patentee may file a patent infringement suit and providing opportunities for third parties to challenge any issued patent in the USPTO. This applies to all of our U.S. patents, even those
13
issued before March 16, 2013. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in U.S. federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. The AIA and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
Additionally, the U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
Any inability of us to protect our competitive advantage with regard to any of our product candidates may prevent us from successfully monetizing such product candidate and this could materially adversely affect our business, prospects, financial condition and results of operations.
We enjoy only limited geographical protection with respect to certain patents and may face difficulties in certain jurisdictions, which may diminish the value of intellectual property rights in those jurisdictions.
International applications under the Patent Cooperation Treaty, or PCT, are usually filed within twelve months after the priority filing. Based on the PCT filing, national and regional patent applications may be filed in additional jurisdictions where we believe our product candidates may be marketed. We have so far not filed for patent protection in all national and regional jurisdictions where such protection may be available. In addition, we may decide to abandon national and regional patent applications before grant. Finally, the grant proceeding of each national/regional patent is an independent proceeding which may lead to situations in which applications might in some jurisdictions be refused by the relevant patent offices, while granted by others. It is also quite common that depending on the country, the scope of patent protection may vary for the same product candidate or technology.
Competitors may use our and our licensors’ or collaboration partners’ technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we and our licensors or collaboration partners have patent protection, but enforcement is not as strong as that in the United States and the European Union. These products may compete with our product candidates, and our and our licensors’ or collaboration partners’ patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws in the United States and the European Union, and companies have encountered significant difficulties in protecting and defending such rights in such jurisdictions. If we or our licensors encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished and we may face additional competition from others in those jurisdictions.
Some countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, some countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we or any of our licensors is forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be impaired and our business, results of operations and financial condition may be adversely affected.
Proceedings to enforce our and our licensors’ or collaboration partners’ patent rights in foreign jurisdictions could result in substantial costs and divert our and our licensors’ or collaboration partners’ efforts and attention from other aspects of our business, could put our and our licensors’ or collaboration partners’ patents at risk of being invalidated or interpreted narrowly and our and our licensors’ or collaboration partners’ patent applications at risk of not issuing and could provoke third parties to assert claims against us or our licensors or collaboration partners. We or our licensors or collaboration partners may not prevail in any lawsuits that we or our licensors or collaboration partners initiate and the damages or other remedies awarded, if any, may not be commercially meaningful.
Litigation or other proceedings or third-party claims of intellectual property infringement could require us to spend significant time and money and could prevent us from selling our products or affect our stock price.
Our commercial success will depend in part on not infringing the patents or copyrights, or otherwise violating the other proprietary rights of others. Significant litigation regarding patent rights and copyright rights occur in our industry. Our competitors around the globe, many of which have substantially greater resources and have made substantial investments in patent portfolios and competing technologies, may have applied for or obtained or may in the future apply for and obtain, patents that will prevent, limit or otherwise
14
interfere with our ability to make, use and sell our products. In addition, patent applications in Europe and elsewhere can be pending for many years before issuance, or unintentionally abandoned patents or applications can be revived, so there may be applications of others now pending or recently revived patents of which we are unaware. These applications may later result in issued patents, or the revival of previously abandoned patents, that will prevent, limit or otherwise interfere with our ability to make, use or sell our products. Third parties may, in the future, assert claims that we are employing their proprietary technology without authorization, including claims from competitors or from non-practicing entities that have no relevant product revenue and against whom our own patent portfolio may have no deterrent effect. As we continue to commercialize our products in their current or updated forms, launch new products and enter new markets, we expect competitors may claim that one or more of our products infringe their intellectual property rights as part of business strategies designed to impede our successful commercialization and entry into new markets. The large number of patents, the rapid rate of new patent applications and issuances, the complexities of the technology involved, and the uncertainty of litigation may increase the risk of business resources and management’s attention being diverted to patent litigation. We have, and we may in the future, receive letters or other threats or claims from third parties inviting us to take licenses under, or alleging that we infringe, their patents.
Moreover, we may become party to future adversarial proceedings regarding our patent portfolio or the patents of third parties. Patents may be subjected to opposition, post-grant review or comparable proceedings lodged in various foreign, both national and regional, patent offices. The legal threshold for initiating litigation or contested proceedings may be low, so that even lawsuits or proceedings with a low probability of success might be initiated. Litigation and contested proceedings can also be expensive and time-consuming, and our adversaries in these proceedings may have the ability to dedicate substantially greater resources to prosecuting these legal actions than we can. We may also occasionally use these proceedings to challenge the patent rights of others. We cannot be certain that any particular challenge will be successful in limiting or eliminating the challenged patent rights of the third party.
Any lawsuits resulting from such allegations could subject us to significant liability for damages and invalidate our proprietary rights. Any potential intellectual property litigation also could force us to do one or more of the following:
Any litigation or claim against us, even those without merit, may cause us to incur substantial costs, and could place a significant strain on our financial resources, divert the attention of management from our core business and harm our reputation. If we are found to infringe the intellectual property rights of third parties, we could be required to pay substantial damages (which may be increased up to three times of awarded damages) and/or substantial royalties and could be prevented from selling our products unless we obtain a license or are able to redesign our products to avoid infringement. Any such license may not be available on reasonable terms, if at all, and there can be no assurance that we would be able to redesign our products in a way that would not infringe the intellectual property rights of others. We could encounter delays in product introductions while we attempt to develop alternative methods or products. If we fail to obtain any required licenses or make any necessary changes to our products or technologies, we may have to withdraw existing products from the market or may be unable to commercialize one or more of our products.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position could be harmed.
We rely on copyright, patent, trade secret, and trademark protection as well as confidentiality agreements with our employees, consultants and third parties, and we may in the future rely on additional intellectual property protection, to protect our confidential and proprietary information. In addition to contractual measures, we try to protect the confidential nature of our proprietary information using commonly accepted physical and technological security measures. Such measures may not, for example, in the case of misappropriation of a trade secret by an employee or third party with authorized access, provide adequate protection for our proprietary information. Our security measures may not prevent an employee or consultant from misappropriating our trade secrets and providing them to a competitor, and recourse we take against such misconduct may not provide an adequate remedy to protect our interests fully. Unauthorized parties may also attempt to copy or reverse engineer certain aspects of our products that we consider proprietary. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive and time-consuming, and the outcome is unpredictable. Even though we use commonly accepted security measures, trade secret violations are often a matter of state law, and the criteria for protection of trade secrets can vary among different jurisdictions. In addition, trade secrets may be independently
15
developed by others in a manner that could prevent legal recourse by us. If any of our confidential or proprietary information, such as our trade secrets, were to be disclosed or misappropriated, or if any such information was independently developed by a competitor, our business and competitive position could be harmed.
Third parties may assert ownership or commercial rights to inventions we develop, which could have a material adverse effect on our business.
Third parties may in the future make claims challenging the inventorship or ownership of our intellectual property. Any infringement claims or lawsuits, even if not meritorious, could be expensive and time consuming to defend, divert management’s attention and resources, require us to redesign our products and services, if feasible, require us to pay royalties or enter into licensing agreements in order to obtain the right to use necessary technologies, and/or may materially disrupt the conduct of our business.
In addition, we may face claims by third parties that our agreements with employees, contractors or third parties obligating them to assign intellectual property to us are ineffective or in conflict with prior or competing contractual obligations of assignment, which could result in ownership disputes regarding intellectual property we have developed or will develop and interfere with our ability to capture the commercial value of such intellectual property. Litigation may be necessary to resolve an ownership dispute, and if we are not successful, we may be precluded from using certain intellectual property or may lose our exclusive rights in that intellectual property. Either outcome could harm our business and competitive position.
Third parties may assert that our employees or contractors have wrongfully used or disclosed confidential information or misappropriated trade secrets, which could result in litigation.
We may employ individuals who previously worked with other companies, including our competitors or potential competitors. Although we try to ensure that our employees and contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees or contractors have inadvertently or otherwise used or disclosed intellectual property or personal data, including trade secrets or other proprietary information, of a former employer or other third party. Litigation may be necessary to defend against these claims. If we fail in defending any such claims or settling those claims, in addition to paying monetary damages or a settlement payment, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Risks Related to Regulatory and Other Legal Issues.
The regulatory environment for IVD could change, resulting a new procedure for achieving approvals for various global marketplaces which might adversely affect Virax’s ability to enter various markets.
Changes in the current regulatory framework for IVD can impose additional regulatory burdens on us. In addition, there is currently an on-going review as to acceptance of EU MDR and EU IVDR CE certificates renewed which we cannot be certain of in future developments. As the regulatory framework evolves in the targeted jurisdictions for our current in-development T Cell IVD/Immune response Test under the ViraxImmune brand, we may incur substantial costs to ensure compliance with new or amended laws and regulations. Failure to comply with any of these laws and regulations could result in enforcement actions against us, damage to our reputation, render us unable to commercialize our ViraxImmune product and/or platform in the expected timeframe or at all, and our ability to expand our business and achieve our strategic objectives would be impaired, any of which could have a material adverse effect on our business.
If we fail to comply with the extensive regulations of domestic and international regulatory authorities, sales of our products in new markets and the development and commercialization of any new product candidates could be delayed or prevented.
Our existing tests, as well as new tests, will be subject to extensive government regulations related to development, testing, manufacturing and commercialization in Europe, the United States and other countries before we can sell in these markets. The process of obtaining and complying with the relevant governmental regulatory approvals and regulations is costly, time consuming, uncertain and subject to unanticipated delays. Despite the time and expense exerted, regulatory approval is never guaranteed. We may not be able to obtain the required regulatory approval and market any further products we may develop during the time we anticipate, or at all. We also are subject to the following risks and obligations, among others:
16
In addition, some international jurisdictions, require periodic re-registration. Even if we obtain initial registrations from regulatory bodies, we may lose registration after a periodic review. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion of our products from government funded healthcare programs, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, reputational harm and the curtailment or restructuring of our operations. Defending against any such actions can be costly and time-consuming and may require significant financial resources.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. For example, the provision of benefits or advantages to physicians to induce or encourage the prescription, recommendation, endorsement, purchase, supply, order or use of medicinal products is generally not permitted in the countries that form part of the European Union. Some European Union Member States have enacted laws explicitly prohibiting the provision of these types of benefits and advantages to induce or reward improper performance generally, and the United Kingdom has enacted such laws through the Bribery Act 2010. Infringements of these laws can result in substantial fines and imprisonment. EU Directive 2001/83/EC, which is the EU Directive governing medicinal products for human use, further provides that, where medicinal products are being promoted to persons qualified to prescribe or supply them, no gifts, pecuniary advantages or benefits in kind may be supplied, offered or promised to such persons unless they are inexpensive and relevant to the practice of medicine or pharmacy. This provision has been transposed into the Human Medicines Regulations 2012 and so remains applicable in the UK despite its departure from the EU. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business.
If we or our suppliers fail to comply with ongoing regulatory requirements, or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any product for which we obtain marketing approval in the United Kingdom or in international jurisdictions, along with the manufacturing processes, post-approval clinical data and promotional activities for such product, will be subject to continual review and periodic inspections by the relevant regulatory bodies. Furthermore, our suppliers may be subject to similar regulatory oversight, and may not currently be or may not continue to be in compliance with applicable regulatory requirements. Failure by us or one of our suppliers to comply with statutes and regulations administered by the relevant regulatory bodies, or failure to take adequate action in response to any observations, could result in, among other things, any of the following enforcement actions:
If any of these actions were to occur, it could harm our reputation and could cause our product sales and profitability to suffer.
Any regulatory approval of a product may also be subject to limitations on the indicated uses for which the product may be marketed. If the FDA or another regulatory body determines that our promotional materials, training or other activities constitute promotion of an unapproved use, it could request that we cease or modify our training or promotional materials or subject us to regulatory enforcement
17
actions. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our training or promotional materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under applicable statutory authorities, such as laws prohibiting false claims for reimbursement. The following are significant regulations that are currently applicable and could also be applicable to our products due to our target markets:
European Union Regulations
The European In-Vitro Diagnostic Regulation (IVDR 2017/746) (“IVDR”) introduced a new risk-based classification system and requirements for conformity assessments. Products self-certified placed on the market before May 22, 2022 under the European In-Vitro Diagnostic Devices Directive (IVDD 98/79/EC) (“IVDD”) may remain on the market until the following dates, afterward they will require the involvement of a Notified Body under the IVDR for the first time:
IVD manufacturers may only rely on the transitional provisions above provided that: (i) the devices continue to comply with applicable requirements imposed by the IVDD; (ii) they respect the IVDR requirements relating to post-market surveillance, market surveillance, vigilance, registration of economic operators and devices from May 26, 2022 in place of the corresponding requirements in the IVDD; and (iii) no significant changes are made in the design and intended purpose of the devices during the transitional period.
CE Marking is required for all IVD devices sold in Europe. CE Marking indicates that an IVD device complies with the IVDR and that the device may be legally commercialized in the EU.
It should be appreciated that there is a severe shortage of capacity of Notified Bodies to assess all IVDs that will require Notified Body certification under the IVDR, and that it is widely recognized that applications for assessment by the Notified Bodies may be subject to significant delays.
United Kingdom Regulations
The UK’s withdrawal from the EU will have major ramifications for IVD manufacturers that will, among other things have to follow new procedures that will apply in the UK including appointment of a UK Responsible Person rather than relying on European Authorized Representatives to manage their compliance efforts in the UK.
IVDs compliant with the EU in vitro diagnostic medical devices regulation (EU IVDD) can be placed on the Great Britain market up until the 30 June 2030. We intend to use the recognized CE marks that we will apply with the European Union for our medical device product, namely our current in development T cell IVD/Immune response Test under the ViraxImmune brand. After which, we will apply with the UK Medicine and Healthcare Products Regulatory Agency for a UK Conformity Assessed mark.
U.S. Regulations
We consider the United States as a target market with significant potential. As such, United States regulations will be applicable to our products once we market our products in the United States. In the United States, the FDA regulates the sale or distribution of medical devices, including but not limited to, IVD test kits. IVD products are subject to regulation by the FDA as medical devices to the extent that they are intended for use in the diagnosis, treatment, mitigation or prevention of disease or other conditions. They are subject to premarket review and post market controls which will differ depending on how the FDA classifies a specific IVD.
The information that must be submitted to the FDA in order to obtain clearance or approval to market a new medical device varies depending on how the medical device is classified by the FDA. Medical devices are classified into one of three risk-based classes depending on the controls deemed by the FDA to be necessary to reasonably ensure their safety and effectiveness. Class I devices are considered the lowest risk and are subject to general controls, including labeling requirements, and adherence to the FDA’s quality system regulations, or QSRs, which are device-specific current good manufacturing practices. Class II (intermediate risk) devices may be subject to premarket notification, or may be 510(k)-exempt, and are generally subject to QSRs, general controls and sometimes special controls, including performance standards and post-market surveillance. Class III (highest risk) devices are subject to most of the previously identified requirements as well as to pre-market approval. Class I devices are exempt from premarket review; most Class II devices require 510(k) clearance, and all Class III devices must receive premarket approval before they can be sold in the United
18
States. The payment of a user fee, which is typically adjusted annually, to the FDA is usually required when a 510(k) notice or premarket approval application is submitted.
510(k) Premarket Notification.
A 510(k) premarket notification requires the sponsor to demonstrate that a medical device is substantially equivalent to another marketed device, termed a “predicate device,” that is legally marketed in the United States and for which a premarket approval was not required. A device is substantially equivalent to a predicate device if it has the same intended use and technological characteristics as the predicate; or has the same intended use but different technological characteristics, where the information submitted to the FDA does not raise new questions of safety and effectiveness and demonstrates that the device is at least as safe and effective as the legally marketed device.
If the FDA believes that the device is not substantially equivalent to a predicate device, it will issue a “Not Substantially Equivalent” (NSE) determination and designate the device as a Class III device, which will require the submission and approval of a PMA before the new device may be marketed. A person who receives an NSE determination in response to a 510(k) submission may, within 30 days of receipt of the NSE determination, submit a de novo request for the FDA to make a risk-based evaluation for classification of the device into Class I or II. Devices that are classified through the de novo process may be marketed and used as predicates for future 510(k) submissions. The FDA continues to reevaluate the 510(k) pathway and process and the de novo process, and has taken what it describes as a risk-based approach to develop innovative regulatory policy to propose a more “contemporary” approach. In October 2017, the FDA published a final guidance entitled, “De Novo Classification Process (Evaluation of Automatic Class III Designation),” and in December 2018, the FDA published a proposed rule which if finalized is intended to provide structure, clarity and transparency on the de novo classification process. In January 2021, it also published a final guidance entitled “Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program.”
Premarket Approval. The PMA process is more complex, costly and time consuming than the 510(k) process. A PMA must be supported by more detailed and comprehensive scientific evidence, including clinical data, to demonstrate the safety and efficacy of the medical device for its intended purpose. If the device is determined to present a “significant risk,” the sponsor may not begin a clinical trial until it submits an investigational device exemption (IDE) to the FDA and obtains approval to begin the trial.
After the PMA is submitted, the FDA has 45 days to make a threshold determination that the PMA is sufficiently complete to permit a substantive review. If the PMA is complete, the FDA will file the PMA. The FDA is subject to a performance goal review time for a PMA that is 180 days from the date of filing, although in practice this review time is longer. Questions from the FDA, requests for additional data and referrals to advisory committees may delay the process considerably. The total process may take several years and there is no guarantee that the PMA will ever be approved. Even if approved, the FDA may limit the indications for which the device may be marketed. The FDA may also request additional clinical data as a condition of approval or after the PMA is approved. Any changes to the medical device may require a supplemental PMA to be submitted and approved before changed medical device may be marketed.
Any products sold by us pursuant to FDA clearances or approvals will be subject to pervasive and continuing regulation by the FDA, including record keeping requirements, reporting of adverse experiences with the use of the device and restrictions on the advertising and promotion of our products. In particular, we may not advertise or otherwise promote our devices for indications, patient populations, or other conditions of use that are not covered by the applicable FDA clearance or approval for the device. Modifications or changes to the device or how it is manufactured may also be separately subject to FDA review and authorization before being commercialized. Device manufacturers are required to register their establishments and list their devices with the FDA and are subject to periodic inspections by the FDA and certain state agencies. Noncompliance with applicable FDA requirements can result in, among other things, warning letters, fines, injunctions, civil penalties, recalls or seizures of products, total or partial suspension of production, refusal of the FDA to grant 510(k) clearance or PMA approval for new devices, withdrawal of 510(k) clearances and/or PMA approvals and criminal prosecution.
As a result of the SARS-CoV-2 pandemic, the US government declared a state of emergency which enabled the FDA to issue emergency use authorizations (EUAs) to provide more timely access to critical medical products (including medicines and tests) that may help during the emergency when there are no adequate, approved, and available alternative options. EUAs are in effect until the emergency declaration ends but can be revised or revoked as FDA considers the needs during the emergency and new data on a product’s safety and effectiveness, or as products meet the criteria to become approved, cleared, or licensed by the FDA. Manufacturers of several types of SARS-CoV-2 assays have been granted EUAs. These authorizations are only intended for the duration of the emergency declaration, and afterwards will be revoked. The FDA has indicated the withdrawal of the EUA process will be done in a controlled ramp down.
Additionally, we may be required to conduct costly post-market testing, and we will be required to report adverse events and malfunctions related to our products. Later discovery of previously unknown problems with our products, including unanticipated adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements may result in restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recalls, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties.
19
Furthermore, the relevant authorities will inspect our facilities and those of our suppliers from time to time to determine whether we are in compliance with regulations relating to the manufacture of diagnostic products, including regulations concerning design, manufacture, testing, quality control, product labeling, distribution, promotion and record-keeping practices. A determination that we are in material violation of such regulations could lead to the imposition of civil penalties, including fines, product recalls, product seizures or, in extreme cases, criminal sanctions.
Canada Regulations
We also consider Canada as a target market with significant potential. Health Canada is the department of the Government of Canada responsible for national health policy, for both professional point-of-care and self-testing, which is similar to over-the-counter EUA authorization in the United States. Health Canada regulates, among other things, the research, development, testing, approval, manufacture, packaging, labeling, storage, recordkeeping, advertising, promotion, distribution, marketing, post-approval monitoring and import and export of pharmaceutical, including biologic, products. The process for obtaining regulatory approvals in Canada, along with subsequent compliance with applicable statutes and regulations, require the expenditure of substantial time and financial resources.
We may potentially be subject to product liability claims.
The testing of our T cell IVD/Immune response Test under the ViraxImmune brand entails an inherent risk of product liability claims. Further, providing clinical testing services entails a risk of claims for errors or omissions made by our laboratory staff. Potential liability claims may exceed the amount of our insurance coverage or may be excluded from coverage under the terms of the policy. As of the date of this report, we obtained a product liability insurance for the testing of the T cell IVD/Immune response Test under the ViraxImmune brand. Although we obtained product liability insurance for the testing of the T cell IVD/Immune response Test under the ViraxImmune brand, if any liability claims arise, it may result in:
Any of these outcomes may have an adverse effect on our consolidated results of operations, financial condition and cash flows, and may increase the volatility of our share price.
Our inadvertent or unintentional failure to comply with complex government regulations concerning privacy of medical and personal information could subject us to fines and adversely affect our reputation.
Privacy regulations around the world limit use or disclosure of protected personal information without written authorization or consent, except for permitted purposes outlined in the privacy regulations. The privacy regulations provide for significant fines and other penalties for wrongful use or disclosure of protected health information, including potential civil and criminal fines and penalties.
We have policies and practices that we believe make us compliant with the privacy regulations. Nevertheless, the documentation and process requirements of the privacy regulations are complex and subject to interpretation. Failure to comply with the privacy regulations could subject us to sanctions or penalties, loss of business and negative publicity.
Internationally, virtually every jurisdiction in which we operate has established its own data security and privacy legal framework with which we or our customers must comply, including the General Data Protection Regulation established in the European Union. We may also need to comply with varying and possibly conflicting privacy laws and regulations in other jurisdictions. As a result, we could face regulatory actions, including significant fines or penalties, adverse publicity and possible loss of business.
A disruption in our computer networks, including those related to cybersecurity, could adversely affect our financial performance.
Cybersecurity refers to the combination of technologies, processes and procedures established to protect information technology systems and data from unauthorized access, attack, or damage. We rely on our computer networks and systems, some of which are managed by third parties, to manage and store electronic information (including sensitive data such as confidential business information and personally identifiable data relating to employees, customers and other business partners), and to manage or support a variety of critical business processes and activities. Cyber-attacks are increasingly more common, including in the health care industry. The regulatory environment surrounding information security and privacy is increasingly demanding, with the frequent imposition of new and changing
20
requirements. Compliance with changes in privacy and information security laws and with rapidly evolving industry standards may result in our incurring significant expense due to increased investment in technology and the development of new operational processes.
We have not experienced any known attacks on our information technology systems that have resulted in any material system failure, accident or security breach to date. However, we may face threats to our networks from unauthorized access, security breaches and other system disruptions. We maintain our information technology systems with safeguard protection against cyber-attacks, including passive intrusion protection, specialist security and virus detection software, use of two factor authentication to access systems and the education and training of staff. However, these safeguards do not ensure that a significant cyber-attack could not occur. Although we have taken steps to protect the security of our information systems and the data maintained in those systems, it is possible that our safety and security measures will not prevent the systems’ improper functioning or damage or the improper access or disclosure of personally identifiable information such as in the event of cyber-attacks.
Security breaches, including physical or electronic break-ins, computer viruses, attacks by hackers and similar breaches can create system disruptions or shutdowns or the unauthorized disclosure of confidential information. If personal information or protected health information is improperly accessed, tampered with or disclosed as a result of a security breach, we may incur significant costs to notify and mitigate potential harm to the affected individuals, and we may be subject to sanctions and civil or criminal penalties if we are found to be in violation of the privacy or security rules or other similar laws protecting confidential personal information. In addition, a cybersecurity breach could hurt our reputation by adversely affecting the perception of customers and potential customers of the security of their orders and personal information, subject us to liability claims or regulatory penalties for compromised personal information and could have a material adverse effect on our business, financial condition and results of operations.
We are subject to the U.K. Bribery Act and other anti-corruption laws, as well as export control laws, customs laws, sanctions laws and other laws governing our operations. If we fail to comply with these laws, we could be subject to civil or criminal penalties, other remedial measures, and legal expenses, which could adversely affect our business, results of operations and financial condition.
Our operations are subject to anti-corruption laws, including the Bribery Act and other anti-corruption laws that apply in countries where we do business. The Bribery Act and these other laws generally prohibit us and our employees and intermediaries from bribing, being bribed or making other prohibited payments to government officials or other persons to obtain or retain business or gain some other business advantage. We and our commercial partners operate in a number of jurisdictions that pose a high risk of potential Bribery Act violations, and we participate in collaborations and relationships with third parties whose actions could potentially subject us to liability under the Bribery Act or local anti-corruption laws. In addition, we cannot predict the nature, scope or effect of future regulatory requirements to which our international operations might be subject or the manner in which existing laws might be administered or interpreted.
We are also subject to other laws and regulations governing our international operations, including regulations administered by the governments of the United Kingdom and authorities in the European Union, including applicable export control regulations, economic sanctions on countries and persons, customs requirements and currency exchange regulations, collectively referred to as the Trade Control laws.
There is no assurance that we will be completely effective in ensuring our compliance with all applicable anti-corruption laws, including the Bribery Act, or other legal requirements, including Trade Control laws. If we violate provisions of the Bribery Act or other anti-corruption laws or Trade Control laws, we may be subject to criminal and civil penalties, disgorgement and other sanctions and remedial measures, and legal expenses, which could have an adverse impact on our business, financial condition, results of operations and liquidity. Likewise, any investigation into or audit of us of any potential violations of the Bribery Act and other anti-corruption laws or Trade Control laws by U.K. or other authorities could subject us to fines or criminal or other penalties, which could have an adverse impact on our reputation, our business, results of operations and financial condition.
Recent developments relating to the United Kingdom’s withdrawal from the European Union could adversely affect us.
The recent withdrawal of the United Kingdom from its membership in the European Union, or EU, often referred to as “Brexit”, could lead to legal and regulatory uncertainty in the United Kingdom and may lead to the United Kingdom and European Union adopting divergent laws and regulations, including those related to the pricing of prescription pharmaceuticals, as the United Kingdom determines which European Union laws to replicate or replace. If the United Kingdom were to significantly alter its regulations affecting the pricing of prescription pharmaceuticals, we could face significant new costs. As a result, Brexit could impair our ability to transact business in the European Union and the United Kingdom.
The United Kingdom and the EU have signed a EU-UK Trade and Cooperation Agreement, or TCA, which became provisionally applicable on January 1, 2021 and will become formally applicable once ratified by both the United Kingdom and the EU. This agreement provides details on how some aspects of the United Kingdom and EU’s relationship will operate going forwards however there are still many uncertainties. The uncertainty concerning the United Kingdom’s legal, political and economic relationship with the European Union may be a source of instability in the international markets, create significant currency fluctuations, and/or otherwise
21
adversely affect trading agreements or similar cross-border co-operation arrangements (whether economic, tax, fiscal, legal, regulatory or otherwise).
These developments, or the perception that any of them could occur, have had, and may continue to have, a significant adverse effect on global economic conditions and the stability of global financial markets, and could significantly reduce global market liquidity and limit the ability of key market participants to operate in certain financial markets. In particular, it could also lead to a period of considerable uncertainty in relation to the United Kingdom financial and banking markets, as well as on the regulatory process in Europe. Asset valuations, currency exchange rates and credit ratings may also be subject to increased market volatility.
If other EU Member States pursue withdrawal, barrier-free access among the European Economic Area, or EEA, overall could be diminished or eliminated. The long-term effects of Brexit will depend on how the terms of the TCA take effect in practice and any further agreements (or lack thereof) between the United Kingdom and the EU. Such a withdrawal from the EU is unprecedented, and it is unclear how the UK access to the European single market for goods, capital, services and labor within the EU, and the wider commercial, legal and regulatory environment, will impact our United Kingdom operations.
We may also face new regulatory costs and challenges that could have an adverse effect on our operations and development programs. For example, the United Kingdom will lose the benefits of global trade agreements negotiated by the EU on behalf of its members, which may result in increased trade barriers that could make our doing business in the EU and the EEA more difficult. There may continue to be economic uncertainty surrounding the consequences of Brexit, which could adversely affect our financial condition, results of operations, cash flows and market price of our securities.
We are exposed to unanticipated changes in tax laws and regulations, adjustments to our tax provisions, exposure to additional tax liabilities, or forfeiture of our tax assets.
The determination of our provision for income taxes and other tax liabilities requires significant judgment, including the adoption of certain accounting policies and our determination of whether our deferred tax assets are, and will remain, tax effective. We cannot guarantee that our interpretation or structure will not be questioned by the relevant tax authorities, or that the relevant tax laws and regulations, or the interpretation thereof, including through tax rulings, by the relevant tax authorities, will not be subject to change. Any adverse outcome of such a review may lead to adjustments in the amounts recorded in our financial statements and could have a materially adverse effect on our operating results and financial condition.
We are subject to laws and regulations on tax levies and other charges or contributions in different countries, including transfer pricing and tax regulations for the compensation of personnel and third parties. Dealings between current and former group companies as well as additional companies that may form part of our group in the future are subject to transfer pricing regulations, which may be subject to change and could affect us. Compliance with these laws and regulations will be more challenging as we expand our international operations, including in connection with potential approvals of our product candidates in Europe, the United States and elsewhere.
Our effective tax rates could be adversely affected by changes in tax laws, treaties and regulations, both internationally and domestically, or the interpretation thereof by the relevant tax authorities, including changes to the patent income deduction, possible changes to the corporate income tax base, wage withholding tax incentive for qualified research and development personnel in the United Kingdom and other tax incentives and the implementation of new tax incentives such as the innovation deduction. An increase of the effective tax rates could have an adverse effect on our business, financial position, results of operations and cash flows.
In addition, we may not be able to use, or changes in tax regulations may affect the use of, certain unrecognized tax assets or credits that we have built over the years. In general, some of these tax losses carry forwards may be forfeited in whole, or in part, as a result of various transactions, or their utilization may be restricted by statutory law in the relevant jurisdiction. Any corporate reorganization by us or any transaction relating to our shareholding structure may result in partial or complete forfeiture of tax loss carry forwards. The tax burden would increase if profits, if any, could not be offset against tax loss carry forwards.
Risk Related to our Corporate Structure
We may rely on dividends and other distributions on equity paid by our subsidiaries to fund any cash and financing requirements we may have, and any limitation on the ability of our subsidiaries to make payments to us could have a material adverse effect on our ability to conduct our business.
Virax Biolabs is a holding company incorporated in the Cayman Islands, and we may rely on dividends and other distributions on equity paid by our subsidiaries for our cash and financing requirements, including the funds necessary to pay dividends and other cash distributions to our shareholders and service any debt we may incur. If any of our subsidiaries incurs debt on its own behalf in the future, the instruments governing the debt may restrict its ability to pay dividends or make other distributions to us.
Under the current practice of the Inland Revenue Authority of Singapore, no tax is payable in Singapore in respect of dividends paid by us. Any limitation on the ability of our SingaporeCo to pay dividends or make other distributions to us could materially and adversely
22
limit our ability to grow, make investments or acquisitions that could be beneficial to our business, pay dividends, or otherwise fund and conduct our business. Shareholders of a Cayman company will not be subject to any income, withholding or capital gains taxes in the Cayman Islands with respect to the holding of their shares in a Cayman company and dividends received on those shares, nor will they be subject to any estate or inheritance taxes in the Cayman Islands. There are no foreign exchange controls in the Cayman Islands. Under the Companies Act (Revised) of the Cayman Islands (the "Cayman Companies Act"), a Cayman company may declare and pay a dividend to shareholders from time to time out of the profits or out of the share premium account, provided that Virax Biolabs shall be able to pay its debts as they fall due in the ordinary course of business.
Any limitation on the ability of Virax Biolabs or UK subsidiaries to pay dividends or make other distributions to us could materially and adversely limit our ability to grow, make investments or acquisitions that could be beneficial to our business, pay dividends, or otherwise fund and conduct our business.
Risks related to Singapore
Developments in the social, political, regulatory and economic environment in the countries where we operate, may have a material and adverse impact on us.
Our business, prospects, financial condition and results of operations may be adversely affected by social, political, regulatory and economic developments in countries in which we operate. Such political and economic uncertainties include, but are not limited to, the risks of war, terrorism, nationalism, nullification of contract, changes in interest rates, imposition of capital controls and methods of taxation. For example, we used to have considerable operations in Singapore, and negative developments in Singapore’s socio-political environment may adversely affect our business, financial condition, results of operations and prospects. Although the overall economic environment in Singapore and other countries including the United States and Europe where we operate appear to be positive, there can be no assurance that this will continue to prevail in the future. We are considering closing our Singapore operations due to limited operations in Singapore since our IPO in 2022.
Disruptions in the international trading environment may seriously decrease our international sales.
The success and profitability of our international activities depend on certain factors beyond our control, such as general economic conditions, labor conditions, political stability, macro-economic regulating measures, tax laws, import and export duties, transportation difficulties, fluctuation of local currency and foreign exchange controls of the countries in which we sell our services, as well as the political and economic relationships among the jurisdictions where we source products and jurisdictions where our clients’ customers are located. As a result, our sales will continue to be vulnerable to disruptions in the international trading environment, including adverse changes in foreign government regulations, political unrest and international economic downturns. Any disruptions in the international trading environment may affect the demand for our products, which could impact our business, financial condition and results of operations.
Risks Related to Doing Business in China and Hong Kong
A downturn in the Hong Kong, China or global economy, and economic and political policies of China could materially and adversely affect our business and financial condition.
A part of our operations is located in Hong Kong and China. Accordingly, our business, prospects, financial condition and results of operations may be influenced to a significant degree by political, economic and social conditions in Hong Kong and China generally and by continued economic growth in Hong Kong and China as a whole. The Chinese economy differs from the economies of most developed countries in many respects, including the amount of government involvement, level of development, growth rate, control of foreign exchange and allocation of resources. While the Chinese economy has experienced significant growth over the past decades, growth has been uneven, both geographically and among various sectors of the economy. The Chinese government has implemented various measures to encourage economic growth and guide the allocation of resources. Some of these measures may benefit the overall Chinese economy, but may have a negative effect on us.
Economic conditions in Hong Kong and China are sensitive to global economic conditions. Any prolonged slowdown in the global or Chinese economy may affect potential clients’ confidence in financial markets as a whole and have a negative impact on our business, results of operations and financial condition. Additionally, continued turbulence in the international markets may adversely affect our ability to access the capital markets to meet liquidity needs.
The Hong Kong legal system embodies uncertainties which could limit the legal protections available to us.
Hong Kong is a Special Administrative Region of the PRC. Following British colonial rule from 1842 to 1997, China assumed sovereignty under the “one country, two systems” principle. The Hong Kong Special Administrative Region’s constitutional document, the Basic Law, ensures that the current political situation will remain in effect for 50 years. Hong Kong has enjoyed the freedom to function in a high degree of autonomy for its affairs, including currencies, immigration and customs, independent judiciary system and
23
parliamentary system. On July 14, 2020, the United States signed an executive order to end the special status enjoyed by Hong Kong post-1997. As the autonomy currently enjoyed were compromised, it could potentially impact Hong Kong’s common law legal system and may in turn bring about uncertainty in, for example, the enforcement of our contractual rights. This could, in turn, materially and adversely affect our business and operation. Accordingly, we cannot predict the effect of future developments in the Hong Kong legal system, including the promulgation of new laws, changes to existing laws or the interpretation or enforcement thereof, or the pre-emption of local regulations by national laws. These uncertainties could limit the legal protections available to us, including our ability to enforce our agreements with our clients.
Uncertainties with respect to the PRC legal system, including uncertainties regarding the enforcement of laws, and sudden or unexpected changes in laws and regulations in China could adversely affect us.
A part of our operations is located in China, and thus, Shanghai Xitu is governed by PRC laws and regulations. PRC companies are generally subject to laws and regulations applicable to foreign investments in China and, in particular, laws and regulations applicable to wholly foreign-owned enterprises. The PRC legal system is a civil law system based on written statutes. Unlike the common law system, prior court decisions may be cited for reference but have limited precedential value.
In 1979, the PRC government began to promulgate a comprehensive system of laws, rules and regulations governing economic matters in general. The overall effect of legislation over the past four decades has significantly enhanced the protections afforded to various forms of foreign investment in China. However, China has not developed a fully integrated legal system, and recently enacted laws, rules and regulations may not sufficiently cover all aspects of economic activities in China or may be subject to significant degrees of interpretation by PRC regulatory agencies. In particular, because these laws, rules and regulations are relatively new, and because of the limited number of published decisions and the nonbinding nature of such decisions, and because the laws, rules and regulations often give the relevant regulator significant discretion in how to enforce them, the interpretation and enforcement of these laws, rules and regulations involve uncertainties and can be inconsistent and unpredictable. In addition, the PRC legal system is based in part on government policies and internal rules, some of which are not published on a timely basis or at all, and which may have a retroactive effect. As a result, we may not be aware of our violation of these policies and rules until after the occurrence of the violation.
Any administrative and court proceedings in China may be protracted, resulting in substantial costs and diversion of resources and management attention. Since PRC administrative and court authorities have significant discretion in interpreting and implementing statutory and contractual terms, it may be more difficult to evaluate the outcome of administrative and court proceedings and the level of legal protection we enjoy than in more developed legal systems. These uncertainties may impede our ability to enforce the contracts we have entered into and could materially and adversely affect our business, financial condition and results of operations.
In addition, the Opinions jointly issued by the General Office of the Central Committee of the Communist Party of China and the General Office of the State Council on July 6, 2021 called for strengthened regulation over illegal securities activities and supervision of overseas listings by China-based companies and propose to take effective measures. On September 8, 2022, the Supreme People's Court, the Supreme People's Procuratorate, the Ministry of Public Security and the China Securities Regulatory Commission jointly promulgated the Circular on Issuing the Typical Cases of Strictly Cracking down on Securities Crimes in Accordance with Law, releasing five Typical Cases of Securities Crimes for reference at the time of handling cases.
On December 28, 2021, the Cyberspace Administration of China, or the CAC, published the Measures for Cybersecurity Review which became effective on February 15, 2022, which required that any “network platform operator” controlling personal information of no less than one million users which seeks to list on a foreign stock exchange should also be subject to cybersecurity review. The PRC Data Security Law, which took effect on September 1, 2021, imposes data security and privacy obligations on entities and individuals that carry out data activities, provides for a national security review procedure for data activities that may affect national security and imposes export restrictions on certain data and information. On August 20, 2021, the Standing Committee of the People’s Congress promulgated the PRC Personal Information Protection Law (the “PIPL”), which took effect on November 1, 2021. The PIPL sets out the regulatory framework for handling and protection of personal information and transmission of personal information to overseas. Shanghai Xitu is not a network platform operator, nor does it conduct data activities that may affect national security or hold personal information of more than one million users or conduct any cross-border transfer of personal information from China to overseas. Thus, we do not believe we fall in the “operators of critical information infrastructure” and we are not subject to PRC cybersecurity review. However, the Measures for Cybersecurity Review (2021 version), the Data Security Law and the PIPL were recently adopted and remain unclear on how it will be interpreted, amended and implemented by the relevant PRC governmental authorities.
On February 17, 2023, the State Council and the China Securities Regulatory Commission ("CSRC") promulgated the Trial Administrative Measures of Overseas Securities Offering and Listing by Domestic Companies (the “Administrative Measures”), which took effect on March 31, 2023.
Pursuant to the Article 14 of the Administrative Measures, domestic enterprises that directly offer or list securities on an overseas stock exchange shall file with the CSRC. Domestic enterprise that indirectly offer or list securities on an overseas stock exchange, the issuer shall designate a major domestic operating entity as the domestic responsible person who shall file with the CSRC.
24
We have not “directly” offered securities overseas (as Shanghai Xitu is not the issuer of the listed securities on an overseas stock exchange and does not meet the thresholds discussed below).
According to the Article 15 of the Administrative Measures, if the issuer meets the following conditions, it shall be deemed as an “indirect” overseas offering and listing of a domestic enterprise:
Based on the above mentioned Administrative Measures at IPO, given that Shanghai Xitu does not directly offer or list securities on an overseas stock exchange, and the operating revenue, total profit, total assets or net assets of the Shanghai Xitu for the financial year before our listing accounted for less than 50% of the Virax Company's audited consolidated financial statements. Shanghai Xitu is primarily engaged in procurement, the main parts of the business activities of the Company are not carried out in the PRC, and none of Shanghai Xitu’s senior managers was a Chinese citizen and only two (2) out of seven (7) have an ordinary residence located in the PRC, the Company's IPO shall not be deemed as a domestic enterprise that indirectly offer or list securities on an overseas stock exchange, nor does it require filing or approvals from the CSRC. However, there can be no assurance that the relevant PRC governmental authorities, including the CSRC, would reach the same conclusion as us, or that the CSRC or any other PRC governmental authorities would not promulgate new rules or new interpretation of current rules (with retrospective effect) to require us to obtain CSRC or other PRC governmental approvals for the Company's IPO.
The PRC government has significant oversight and discretion over the conduct of a PRC company’s business and may intervene with or influence its operations at any time as the government deems appropriate to further regulatory, political and societal goals. The PRC government has recently published new policies that significantly affected certain industries such as the education and internet industries, and we cannot rule out the possibility that it will in the future release regulations or policies regarding any industry that could adversely affect the business, financial condition and results of operations of Shanghai Xitu. Furthermore, the PRC government has also recently indicated an intent to exert more oversight and control over securities offerings and other capital markets activities that are conducted overseas and foreign investment in China-based companies. Any such action, once taken by the PRC government, could significantly limit or completely hinder our ability to offer or continue to offer securities to investors and cause the value of such securities to significantly decline or in extreme cases, become worthless.
Uncertainties regarding the enforcement of laws and the fact that rules and regulations in China can change quickly with little advance notice, along with the risk that the Chinese government may intervene or influence our operations at any time, could result in a material change in our operations, financial performance and/or the value of our ordinary shares or impair our ability to raise money.
The Chinese government exerts substantial influence over the manner in which we must conduct our business activities, and may intervene or influence our operations at any time, or may exert more oversight and control over offerings conducted overseas, which could significantly limit or completely hinder our ability to offer or continue to offer our ordinary shares to investors and could cause the value of our ordinary shares to significantly decline or become worthless.
The Chinese government has exercised and continues to exercise substantial control over virtually every sector of the Chinese economy through regulation and state ownership. While there is no revenue from Shanghai Xitu, our ability to operate Shanghai Xitu in China may be harmed by changes in its laws and regulations, including those relating to taxation, environmental regulations, land use rights, property and other matters. The central or local governments of these jurisdictions may impose new, stricter regulations or interpretations of existing regulations that would require additional expenditures and efforts on our part to ensure our compliance with such regulations or interpretations. Accordingly, government actions in the future, including any decision not to continue to support recent economic reforms and to return to a more centrally planned economy or regional or local variations in the implementation of economic policies, could have a significant effect on economic conditions in China or particular regions thereof, and could require us to divest ourselves of any interest we then hold in Chinese properties.
For example, the Chinese cybersecurity regulator announced on July 2, 2021 that it had begun an investigation of Didi Global Inc. (NYSE: DIDI) and two days later ordered that Didi Global Inc.’s app be removed from smartphone app stores.
As such Shanghai Xitu may be subjected to various government and regulatory interference in the provinces in which they operate. Shanghai Xitu could be subjected to regulations by various political and regulatory entities, including various local and municipal agencies and government sub-divisions. We may incur increased costs necessary to comply with existing and newly adopted laws and regulations or penalties for any failure to comply. If the PRC government initiates an investigation into us at any time alleging violation of cybersecurity laws, anti-monopoly laws, and securities offering rules in China in connection with the Company's IPO or any future
25
offerings, we may have to spend additional resources and incur additional time delays to comply with the applicable rules, and our business operations will be affected materially and any such action could cause the value of our securities to significantly decline or be worthless.
As at the date of this report, management believes that there are no PRC laws and regulations (including the CSRC, the CAC, or any other government entity) in force explicitly requiring that our Company or Shanghai Xitu obtain permission from PRC authorities for the Company's IPO or future offerings or to issue securities to foreign investors (by Virax Biolabs), and our Company or Shanghai Xitu have not received any inquiry, notice, warning, sanction or any regulatory objection to the Company's IPO from any relevant PRC authorities. However, it is uncertain when and whether the Company or Shanghai Xitu will be required to obtain permission from the PRC government for future offerings on U.S. stock exchanges, and even when such permission is obtained, whether it will be denied or rescinded. Any new policies, regulations, rules, actions or laws by the PRC government may subject us to material changes in operations, which could significantly limit or completely hinder our ability to offer or continue to offer securities to investors and cause the value of our securities to significantly decline or become worthless.
The Chinese government may exercise significant oversight and discretion over the conduct of Shanghai Xitu’s business and may intervene in or influence its operations at any time, which could result in a material change in its operations and/or the value of our securities.
The Chinese government has exercised and continues to exercise substantial control over virtually every sector of the Chinese economy through regulation and state ownership. Our ability to operate through our PRC subsidiary, Shanghai Xitu, may be harmed by changes in its laws and regulations, including those relating to taxation, environmental regulations, land use rights, property and other matters. The central or local governments of these jurisdictions may impose new, stricter regulations or interpretations of existing regulations that would require additional expenditures and efforts on our part to ensure our compliance with such regulations or interpretations. Accordingly, government actions in the future, including any decision not to continue to support recent economic reforms and to return to a more centrally planned economy or regional or local variations in the implementation of economic policies, could have a significant effect on economic conditions in China or particular regions thereof, and could require us to divest ourselves of any interest we then hold in Chinese properties.
For example, the Chinese cybersecurity regulator announced on July 2, 2021 that it had begun an investigation of Didi Global Inc. (NYSE: DIDI) and two days later ordered that Didi Global Inc.’s app be removed from smartphone app stores. On July 24, 2021, the General Office of the Communist Party of China Central Committee and the General Office of the State Council jointly released the Guidelines for Further Easing the Burden of Excessive Homework and Off-campus Tutoring for Students at the Stage of Compulsory Education, pursuant to which foreign investment in such firms via mergers and acquisitions, franchise development, and variable interest entities are banned from this sector.
As such, Shanghai Xitu’s business segments may be subject to various government and regulatory interference in the provinces in which it operates. Shanghai Xitu could be subject to regulations by various political and regulatory entities, including various local and municipal agencies and government sub-divisions, and these regulations may be interpreted and applied inconsistently by different agencies or authorities. The PRC Target Company may incur increased costs necessary to comply with existing and newly adopted laws and regulations or penalties for any failure to comply, and such compliance or any associated inquiries or investigations or any other government actions may:
The promulgation of new laws or regulations, or the new interpretation of existing laws and regulations, in each case that restrict or otherwise unfavorably may impact the ability or way Shanghai Xitu may conduct its business and could require it to change certain aspects of its business to ensure compliance, which could increase costs, require us to obtain more licenses, permits, approvals or certificates, or subject it to additional liabilities. As such, Shanghai Xitu’s operations could be adversely affected, directly or indirectly, by existing or future PRC laws and regulations relating to its business or industry, which could result in a material adverse change in the value of our securities, potentially rendering it worthless. As a result, both you and us face uncertainty about future actions by the PRC government that could significantly affect our ability to offer or continue to offer securities to investors and cause the value of our securities to significantly decline or be worthless.
26
Changes in China’s economic, political or social conditions or government policies could have a material adverse effect on our Company’s business and results of operations we may pursue in the future.
A part of our operations is located in China and Hong Kong, and thus, or business, prospects, financial condition and results of operations may be influenced to a significant degree by political, economic and social conditions in China generally and by continued economic growth in China as a whole. Policies, regulations, rules, and the enforcement of laws of the PRC government can have significant effects on economic conditions in the PRC and the ability of businesses to operate profitably. Shanghai Xitu’s, HKco’s, and ViraxImmune T cell’s ability to operate profitably in the PRC and Hong Kong may be adversely affected by changes in policies by the PRC government, including changes in laws, regulations or their interpretation, particularly those dealing with the Internet, including censorship and other restriction on material which can be transmitted over the Internet, security, intellectual property, money laundering, taxation and other laws that affect our PRC and Hong Kong subsidiaries’ ability to operate its business.
Any actions by the PRC government to exert more oversight and control over offerings (including businesses whose primary operations are in Hong Kong) that are conducted overseas and/or foreign investments in Hong Kong- or PRC-based issuers could significantly limit or completely hinder our ability to offer or continue to offer securities to investors and cause the value of our securities to significantly decline or be worthless.
PRC regulation of loans to and direct investment in PRC entities by offshore holding companies and governmental control of currency conversion may delay us from using part of the proceeds of the Company's IPO to make loans or additional capital contributions to our PRC subsidiary, which could materially and adversely affect our liquidity and our ability to fund and expand our business.
Any funds our Company transfers to our Shanghai Xitu, either as a shareholder loan or as an increase in registered capital, are subject to approval by or registration with relevant governmental authorities in China. According to the relevant PRC regulations on FIEs in China, capital contributions to our PRC subsidiary are subject to registration with the SAMR (or its local branches) and filing with the Ministry of Commerce of the PRC, or the MOFCOM, or its local branches and (if applicable) registration with other relevant governmental authorities in China. In addition, (a) any foreign loan procured by our PRC subsidiary is required to be registered with SAFE or its local branches, and (b) our PRC subsidiary may not procure loans which exceed the statutory amount as approved by the MOFCOM or its local branches. Any medium-or long-term loan to be provided by us to our PRC subsidiary must be approved by the National Development and Reform Commission, or NDRC and the SAFE or its local branches. We may not obtain these government approvals or complete such registrations on a timely basis, with respect to future capital contributions or foreign loans by us to our PRC subsidiary. If we fail to receive such approvals or complete such registration, our ability to use part of the proceeds of the Company's IPO and to capitalize our PRC operations may be negatively affected, which could materially and adversely affect our liquidity and our ability to fund and expand our business.
In 2008, SAFE promulgated the Circular on the Relevant Operating Issues Concerning the Improvement of the Administration of the Payment and Settlement of Foreign Currency Capital of Foreign-Invested Enterprises, or SAFE Circular 142. SAFE Circular 142 regulates the conversion by FIEs of foreign currency into Renminbi by restricting the usage of converted Renminbi. SAFE Circular 142 provides that any Renminbi capital converted from registered capital in foreign currency of FIEs may only be used for purposes within the business scopes approved by PRC governmental authority and such Renminbi capital may not be used for equity investments within China unless otherwise permitted by PRC law. In addition, the SAFE strengthened its oversight of the flow and use of Renminbi capital converted from registered capital in foreign currency of FIEs. The use of such Renminbi capital may not be changed without SAFE approval, and such Renminbi capital may not in any case be used to repay Renminbi loans if the proceeds of such loans have not been utilized. On July 4, 2014, SAFE issued the Circular of the SAFE on Relevant Issues Concerning the Pilot Reform in Certain Areas of the Administrative Method of the Conversion of Foreign Exchange Funds by Foreign-invested Enterprises, or SAFE Circular 36, which launched the pilot reform of administration regarding conversion of foreign currency registered capitals of FIEs in 16 pilot areas. According to SAFE Circular 36, some of the restrictions under SAFE Circular 142 will not apply to the settlement of the foreign exchange capitals of an ordinary FIE in the pilot areas, and such FIE is permitted to use Renminbi converted from its foreign-currency registered capital to make equity investments in the PRC within and in accordance with the authorized business scope of such FIEs, subject to certain registration and settlement procedure as set forth in SAFE Circular 36. On March 30, 2015, the SAFE promulgated the Circular on Reforming the Management Approach Regarding the Foreign Exchange Capital Settlement of Foreign-Invested Enterprises, or SAFE Circular 19. SAFE Circular 19 took effect as of June 1, 2015 and superseded SAFE Circular 36 and SAFE Circular 142 on the same date. SAFE Circular 19 launched a nationwide reform of the administration of the settlement of the foreign exchange capitals of FIEs and allows FIEs to settle their foreign exchange capital at their discretion, but continues to prohibit FIEs from using the Renminbi fund converted from their foreign exchange capitals for expenditure beyond their business scopes, providing entrusted loans or repaying loans between non-financial enterprises. Violations of these Circulars could result in severe monetary or other penalties. SAFE Circular 19 may significantly limit our ability to use Renminbi converted from part of the net proceeds of the Company's IPO to fund the establishment of new entities in China by our subsidiary, to invest in or acquire any other PRC companies through our PRC subsidiary, or to establish variable interest entities in the PRC, which may materially and adversely affect our business, financial condition and results of operations. In light of the various requirements imposed by PRC regulations on loans to and direct investment
27
in PRC entities by offshore holding companies, we cannot assure you that we will be able to complete the necessary registration or obtain the necessary approval on a timely basis, or at all. If we fail to complete the necessary registration or obtain the necessary approval, our ability to make loans or equity contributions to our PRC subsidiary may be negatively affected, which could materially and adversely affect our PRC subsidiary’ liquidity and its ability to fund its working capital and expansion projects and meet its obligations and commitments.
Restrictions on currency exchange may limit our ability to utilize our revenues effectively.
Some of our cash are denominated in Renminbi. The Renminbi is currently convertible under the “current account,” which includes dividends, trade and service-related foreign exchange transactions, but not under the “capital account,” which includes foreign direct investment and loans, including loans we may secure from our onshore subsidiary, Shanghai Xitu. Currently, our Shanghai subsidiary may purchase foreign currency for settlement of “current account transactions,” including payment of dividends to us, without the approval of SAFE by complying with certain procedural requirements. However, the relevant PRC governmental authorities may limit or eliminate our ability to purchase foreign currencies in the future for current account transactions. As we have some operations in PRC, we expect a portion of our cash will be denominated in Renminbi, any existing and future restrictions on currency exchange may limit our ability to utilize our Renminbi to fund our business activities outside of the PRC or pay dividends in foreign currencies to our shareholders. Foreign exchange transactions under the capital account remain subject to limitations and require approvals from, or registration with, SAFE and other relevant PRC governmental authorities. This could affect our ability to obtain foreign currency through debt or equity financing for our subsidiary.
Risks Related to Our Securities
Our share price may be volatile and may fluctuate.
Like other biotechnology companies, the market price of our ordinary Shares may be volatile. The factors below may also have a material adverse effect on the market price of our ordinary Shares:
In addition, in the past, when the market price of a stock has been volatile, holders of that stock have instituted securities class action litigation against the issuer that issued the stock. If any of our shareholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit and divert the time and attention of our management, which could seriously harm our business.
28
If we fail to meet applicable listing requirements, Nasdaq may delist our ordinary shares from trading, in which case the liquidity and market price of our ordinary shares could decline.
We cannot assure you that we will be able to meet the continued listing standards of Nasdaq in the future. If we fail to comply with the applicable listing standards and Nasdaq delists our ordinary Shares, we and our shareholders could face significant material adverse consequences, including:
The National Securities Markets Improvement Act of 1996, which is a federal statute, prevents or preempts the states from regulating the sale of certain securities, which are referred to as “covered securities.” Although the states are preempted from regulating the sale of our securities, the federal statute does allow the states to investigate companies if there is a suspicion of fraud, and, if there is a finding of fraudulent activity, then the states can regulate or bar the sale of covered securities in a particular case. Further, if we were no longer listed on Nasdaq, our securities would not be covered securities and we would be subject to regulations in each state in which we offer our securities.
We do not intend to pay cash dividends on our ordinary shares in the foreseeable future.
We have never paid dividends on ordinary shares and do not currently anticipate paying any cash dividends on our ordinary shares in the foreseeable future. The holders of our ordinary shares are entitled to such dividends as may be declared by our board of directors. Our articles of association provide that our board of directors may declare and pay dividends if justified by our financial position and permitted by law. Our articles of association also provides that, subject to the Cayman Companies Act, the Company may also by ordinary resolution declare dividends in accordance with the respective rights of the shareholders but no dividend shall exceed the amount recommended by the directors.
Under Cayman Islands law, any payment of dividends would be subject to relevant legislation and our articles of association, which provide that all dividends must be approved by our board of directors and, in some cases, our shareholders, and may only be paid from our distributable profits available for the purpose, determined on an unconsolidated basis.
We are an emerging growth company within the meaning of the Securities Act and may take advantage of certain reduced reporting requirements.
We are an “emerging growth company,” as defined in the JOBS Act, and we may take advantage of certain exemptions from requirements applicable to other public companies that are not emerging growth companies, including, most significantly, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002 for so long as we remain an emerging growth company. As a result, if we elect not to comply with such auditor attestation requirements, our investors may not have access to certain information they may deem important.
The JOBS Act also provides that an emerging growth company does not need to comply with any new or revised financial accounting standards until such date that a private company is otherwise required to comply with such new or revised accounting standards. We do not plan to “opt out” of such exemptions afforded to an emerging growth company. As a result of this election, our financial statements may not be comparable to those of companies that comply with public company effective dates.
We qualify as a foreign private issuer and, as a result, we will not be subject to U.S. proxy rules and will be subject to Exchange Act reporting obligations that permit less detailed and less frequent reporting than that of a U.S. domestic public company.
Upon the closing of the Company's IPO, we report under the Exchange Act as a non-U.S. company with foreign private issuer status. Because we qualify as a foreign private issuer under the Exchange Act, we are exempt from certain provisions of the Exchange Act that are applicable to U.S. domestic public companies, including (i) the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act; (ii) the sections of the Exchange Act requiring insiders to file public reports of their stock ownership and trading activities and liability for insiders who profit from trades made in a short period of time; and (iii) the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q
29
containing unaudited financial and other specified information, or current reports on Form 8-K upon the occurrence of specified significant events. In addition, our officers, directors and principal shareholders are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and the rules thereunder. Therefore, our shareholders may not know on a timely basis when our officers, directors and principal shareholders purchase or sell our ordinary shares. In addition, foreign private issuers are not required to file their annual report on Form 20-F until one hundred twenty (120) days after the end of each fiscal year, while U.S. domestic issuers that are accelerated filers are required to file their annual report on Form 10-K within seventy-five (75) days after the end of each fiscal year. Foreign private issuers also are exempt from Regulation Fair Disclosure, aimed at preventing issuers from making selective disclosures of material information. As a result of the above, you may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
If we lose our status as a foreign private issuer, we would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. We may also be required to make changes in our corporate governance practices in accordance with various SEC and Nasdaq rules. The regulatory and compliance costs to us under U.S. securities laws if we are required to comply with the reporting requirements applicable to a U.S. domestic issuer may be significantly higher than the cost we would incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status would increase our legal and financial compliance costs and would make some activities highly time consuming and costly. We also expect that if we were required to comply with the rules and regulations applicable to U.S. domestic issuers, it would make it more difficult and expensive for us to obtain and maintain directors’ and officers’ liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These rules and regulations could also make it more difficult for us to attract and retain qualified members of our board of directors.
As a foreign private issuer, we are permitted to adopt certain home country practices in relation to corporate governance matters that differ significantly from Nasdaq corporate governance listing standards. These practices may afford less protection to shareholders than they would enjoy if we complied fully with corporate governance listing standards.
As a foreign private issuer, we are permitted to take advantage of certain provisions in the Nasdaq rules that allow us to follow our home country law for certain governance matters. Certain corporate governance practices in our home country, the Cayman Islands, may differ significantly from corporate governance listing standards. We have adopted Cayman Islands practices in lieu of certain requirements of Rule 5635 of the Nasdaq Stock Market LLC Rules which, among others, means we do not have to obtain shareholders' approval for certain dilutive events, such as (i) certain acquisition of stock or assets of another company; (ii) an issuance of shares that will result in a change of control of the company; (iii) the establishment or amendment of certain equity based compensation plans and arrangements; and (iv) certain transactions (other than a public offering) involving issuances of a 20% or more interest or voting power in the company at a price that is less than the minimum price defined therein. As such, our shareholders may be afforded less protection than they would otherwise enjoy under the Nasdaq corporate governance listing standards applicable to U.S. domestic issuers.
We may lose our foreign private issuer status in the future, which could result in significant additional costs and expenses.
As discussed above, we are a foreign private issuer, and therefore, we are not required to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act. The determination of foreign private issuer status is made annually on the last business day of an issuer’s most recently completed second fiscal quarter. We would lose our foreign private issuer status if, for example, more than 50% of our ordinary shares are directly or indirectly held by residents of the United States and we fail to meet additional requirements necessary to maintain our foreign private issuer status. If we lose our foreign private issuer status on this date, we will be required to file with the SEC periodic reports and registration statements on U.S. domestic issuer forms, which are more detailed and extensive than the forms available to a foreign private issuer. We will also have to mandatorily comply with U.S. federal proxy requirements, and our officers, directors and principal shareholders will become subject to the short-swing profit disclosure and recovery provisions of Section 16 of the Exchange Act. In addition, we will lose our ability to rely upon exemptions from certain corporate governance requirements under the Nasdaq rules. As a U.S. listed public company that is not a foreign private issuer, we will incur significant additional legal, accounting and other expenses that we will not incur as a foreign private issuer, and additional accounting, reporting and other expenses in order to maintain a listing on a U.S. securities exchange.
Nasdaq may apply additional and more stringent criteria for our continued listing because our insiders will hold a large portion of our listed securities.
Under Listing Rule 5101, Nasdaq has discretionary authority to deny initial listing, apply additional or more stringent criteria for the initial or continued listing of particular securities, or suspend or delist particular securities based on any event, condition, or circumstance that exists or occurs that makes initial or continued listing of the securities on Nasdaq inadvisable or unwarranted in the opinion of Nasdaq, even though the securities meet all enumerated criteria for initial or continued listing on Nasdaq.
Additionally, Nasdaq has used its discretion to deny initial or continued listing or to apply additional and more stringent criteria in the instances, including but not limited to: (i) where the company engaged an auditor that has not been subject to an inspection by PCAOB, an auditor that PCAOB cannot inspect, or an auditor that has not demonstrated sufficient resources, geographic reach, or experience to
30
adequately perform the company’s audit; (ii) where the company planned a small public offering, which would result in insiders holding a large portion of the company’s listed securities. Nasdaq was concerned that the offering size was insufficient to establish the company’s initial valuation, and there would not be sufficient liquidity to support a public market for the company; and (iii) where the company did not demonstrate sufficient nexus to the U.S. capital market, including having no U.S. shareholders, operations, or members of the board of directors or management. Therefore, we may be subject to the additional and more stringent criteria of Nasdaq for our initial and continued listing.
Failure to comply with anti-corruption and anti-money laundering laws, including the FCPA and similar laws associated with activities outside of the United States, could subject us to penalties and other adverse consequences.
We are subject to the Foreign Corrupt Practices Act of 1977, as amended, 15 U.S.C. §§ 78dd-1, et seq., referred to as the FCPA, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the USA PATRIOT Act, the UK Bribery Act, and possibly other anti-bribery and anti-money laundering laws in countries in which we conduct activities. We face significant risks if we fail to comply with the FCPA and other anti-corruption laws that prohibit companies and their employees and third-party intermediaries from promising, authorizing, offering, or providing, directly or indirectly, improper payments or benefits to foreign government officials, political parties, and private-sector recipients for the purpose of obtaining or retaining business, directing business to any person, or securing any advantage. Any violation of the FCPA, other applicable anti-corruption laws, and anti-money laundering laws could result in whistleblower complaints, adverse media coverage, investigations, loss of export privileges, or severe criminal or civil sanctions, which could have a material adverse effect on our reputation, business, operating results, and prospects. In addition, responding to any enforcement action may result in a significant diversion of management’s attention and resources, significant defense costs, and other professional fees.
We incur significant costs as a result of being a public company, which may materially and adversely affect our business, financial condition and results of operations.
We incur costs associated with corporate governance requirements that are applicable to us as a public company, including rules and regulations of the SEC, under the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, and the Exchange Act, as well as the rules of the Nasdaq. These rules and regulations significantly increase our accounting, legal and financial compliance costs and make some activities more time-consuming and regulations to make it more expensive for us to obtain and maintain directors’ and officers’ liability insurance. As a result, it may be more difficult for us to attract and retain qualified persons to serve on our board of directors or as executive officers. Accordingly, increases in costs incurred as a result of being a publicly traded company may materially and adversely affect our business, financial condition and results of operations.
Securities analysts may not publish favorable research or reports about our business or may publish no information at all, which could cause our stock price or trading volume to decline.
The trading market will be influenced to some extent by the research and reports that industry or financial analysts publish about us and our business. We do not control these analysts. As a newly public company, we may be slow to attract research coverage and the analysts who publish information about our securities will have had relatively little experience with us or our industry, which could affect their ability to accurately forecast our results and could make it more likely that we fail to meet their estimates. In the event we obtain securities or industry analyst coverage, if any of the analysts who cover us provide inaccurate or unfavorable research or issue an adverse opinion regarding our stock price, our stock price could decline. If one or more of these analysts cease coverage of us or fail to publish reports covering us regularly, we could lose visibility in the market, which in turn could cause our stock price or trading volume to decline and result in the loss of all or a part of your investment in us.
Recently introduced economic substance legislation of the Cayman Islands may impact us and our operations.
Pursuant to the International Tax Cooperation (Economic Substance) Act, 2018 of the Cayman Islands, or the ES Act, that came into force on January 1, 2019, a “relevant entity” is required to satisfy the economic substance test set out in the ES Act. A “relevant entity” includes an exempted company incorporated in the Cayman Islands as is Virax Biolabs. There are nine designated "relevant activities" under the ES Act, and for so long as Virax Biolabs is carrying on activities which falls within any of the designated relevant activities, it shall comply with all applicable requirements under the ES Act. If the only business activity that Virax Biolabs carries on is to hold equity participation in other entities and only earns dividends and capital gains, then based on the current interpretation of the ES Act, Virax Biolabs is a “pure equity holding company” and will therefore only be subject to the minimum substance requirements, which require Virax Biolabs to (i) comply with the all applicable requirements under the Cayman Companies Act and (ii) have adequate human resources and adequate premises in the Cayman Islands for holding and managing equity participations in other entities. However, there can be no assurance that Virax Biolabs will not be subject to more requirements under the ES Act. Uncertainties over the interpretation and implementation of the ES Act may have an adverse impact on Virax Biolabs' business and operations.
31
Because we are incorporated under the laws of the Cayman Islands, our executive office is located in United Kingdom and the majority of our executive officers and directors are located outside the United States, you may face difficulties in protecting your interests, and your ability to protect your rights through the U.S. Federal or state courts may be limited.
We are holding company incorporated as an exempted company with limited liability incorporated under the laws of the Cayman Islands and our executive office is located in the United Kingdom. In addition, the majority of our executive officers and directors are located outside of the United States and are nationals or residents of jurisdictions other than the United States, and all or a substantial portion of their assets are located outside of the United States. Mr. James Foster, our Chief Executive Officer, chairman of the board of directors, holds a British Passport and currently resides in Shanghai, China; Mr. Jason Davis, our Chief Financial Officer, is located in the United States and holds a United States passport; Dr. Nigel McCracken, our Chief Operating Officer, holds a British passport and currently resides in the United Kingdom; Mr. Yair Erez, our independent director, holds a British passport and currently resides in the United Kingdom; Mr. Evan Norton, our independent director, holds a United States passport and currently resides in the United States; and Mr. Nelson Haight, our independent director, holds a United States passport and currently resides in United States.
As a result, it may be difficult for investors to effect service of process within the United States upon us or these persons, or to enforce judgments obtained in U.S. courts against us or them, including judgments predicated upon the civil liability provisions of the securities laws of the United States or any state in the United States. A judgment of a United States court for civil liabilities predicated upon the federal securities laws of the United States may not be enforceable in or recognized by the courts of the jurisdictions where our directors and officers reside, and the judicial recognition process may be time-consuming. It may be difficult for you to enforce judgments obtained in U.S. courts based on the civil liability provisions of the U.S. federal securities laws against us and our officers and directors.
We have an U.S. office in Texas to receive service of process with respect to any action brought against us in the state or federal courts of the United States.
Our corporate affairs are governed by our memorandum and articles of association, the Cayman Companies Act and the common law of the Cayman Islands. The rights of shareholders to take action against our directors, actions by our minority shareholders and the fiduciary duties of our directors to us under Cayman Islands law are to a large extent governed by the common law of the Cayman Islands. The common law of the Cayman Islands is derived in part from comparatively limited judicial precedent in the Cayman Islands as well as from the common law of England and Wales, the decisions of whose courts are of persuasive authority, but are not binding, on a court in the Cayman Islands. The rights of our shareholders and the fiduciary duties of our directors under Cayman Islands law are not as clearly established as they would be under statutes or judicial precedent in some jurisdictions in the United States. In particular, the Cayman Islands have a less developed body of securities laws than the United States. Some U.S. states, such as Delaware, have more fully developed and judicially interpreted bodies of corporate law than the Cayman Islands. In addition, Cayman Islands companies may not have standing to initiate a shareholder derivative action in a federal court of the United States.
We have been advised by our Cayman Islands legal counsel that there is uncertainty as to whether the courts of the Cayman Islands would:
There is no statutory enforcement in the Cayman Islands of judgments obtained in the United States, although the courts of the Cayman Islands will in certain circumstances recognize and enforce a foreign judgment, without any re-examination or re-litigation of matters adjudicated upon, provided such judgment:
Subject to the above limitations, in appropriate circumstances, a Cayman Islands court may give effect in the Cayman Islands to other kinds of final foreign judgments such as declaratory orders, orders for performance of contracts and injunctions.
Moreover, while under Delaware law, controlling shareholders owe fiduciary duties to the companies they control and their minority shareholders, under Cayman Islands law, our controlling shareholders do not owe any such fiduciary duties to our company or to our
32
minority shareholders. Accordingly, our controlling shareholders may exercise their powers as shareholders, including the exercise of voting rights in respect of their shares, in such manner as they think fit in most circumstances.
Shareholders of Cayman exempted companies incorporated in the Cayman Islands like us have no general rights under Cayman Islands law to inspect corporate records (other than the memorandum and articles of association, and any special resolutions passed by such companies, and the registers of mortgages and charges of such companies) or to obtain copies of lists of shareholders of these companies. Our directors have discretion under our memorandum and articles of association to determine whether or not, and under what conditions, our corporate records may be inspected by our shareholders, but are not obliged to make them available to our shareholders unless required by the Cayman Companies Act or other applicable law or authorized by the directors or by ordinary resolution. This may make it more difficult for you to obtain the information needed to establish any facts necessary for a shareholder motion or to solicit proxies from other shareholders in connection with a proxy contest.
Certain corporate governance practices in our home country, the Cayman Islands, may differ significantly from corporate governance listing standards. We have adopted Cayman Islands practices in lieu of certain requirements of Rule 5635 of the Nasdaq Stock Market LLC Rules which, among others, means we do not have to obtain shareholders' approval for certain dilutive events, such as (i) certain acquisition of stock or assets of another company; (ii) an issuance of shares that will result in a change of control of the company; (iii) the establishment or amendment of certain equity based compensation plans and arrangements; and (iv) certain transactions (other than a public offering) involving issuances of a 20% or more interest or voting power in the company at a price that is less than the minimum price defined therein.
As a result of all of the above, our public shareholders may have more difficulty in protecting their interests in the face of actions taken by our management, members of our board of directors or controlling shareholders than they would as public shareholders of a company incorporated in the United States.
Item 4. Information on the Company
A. History and Development of the Company.
Our legal name is Virax Biolabs Group Limited, and our commercial name is “Virax Biolabs.” We are an exempted company with limited liability incorporated under the laws of the Cayman Islands. Our principal executive offices are located at BioCity Glasgow, Bo’Ness Road, Newhouse, Lanarkshire, ML1 5UH. Our telephone number is +44 020 7788 7414.
Virax Biolabs is a holding company incorporated in the Cayman Islands. See Item 4.C. - Organizational Structure for the complete subsidiary hierarchy of the Company. In June 2022, Virax Cayman underwent a shareholding restructuring whereby the Company’s authorized share capital became a single class of ordinary shares and all of the then issued shares were re-designated as ordinary shares. In July 2022, the Company completed its IPO and began trading on the Nasdaq under the symbol “VRAX.”
We had $1,164,449 and $178,403 of capital expenditures associated with the purchase of property, plant and equipment, purchases of software, and investment in internally generated software for the year ended March 31, 2024 and 2023, respectively.
We use our website (http://www.viraxbiolabs.com) as a channel of distribution of Company information. The information we post on our website may be deemed material. Accordingly, investors should monitor the website, in addition to following our press releases, SEC filings and public conference calls and webcasts. The contents of our website are not, however, a part of this Annual Report on Form 20-F. In addition, the SEC maintains an internet site that contains reports, proxy and information statements, and other information regarding our filings and can be found at http://www.sec.gov.
Emerging Growth Company Status
As a company with less than US$1.235 billion in revenue for our last fiscal year, we qualify as an “emerging growth company” pursuant to the Jumpstart Our Business Startups Act of 2012, as amended, or the JOBS Act. An emerging growth company may take advantage of specified reduced reporting and other requirements compared to those that are otherwise applicable generally to public companies. These provisions include exemption from the auditor attestation requirement under Section 404 of the Sarbanes-Oxley Act of 2002 in the assessment of the emerging growth company’s internal control over financial reporting. The JOBS Act also provides that an emerging growth company does not need to comply with any new or revised financial accounting standards until such date that a private company is otherwise required to comply with such new or revised accounting standards. Pursuant to the JOBS Act, we have elected to take advantage of the benefits of this extended transition period for complying with new or revised accounting standards. As a result, our operating results and financial statements may not be comparable to the operating results and financial statements of other companies who have adopted the new or revised accounting standards.
We will remain an emerging growth company until the earliest of (i) the last day of the fiscal year during which we have total annual gross revenues of at least US$1.235 billion; (ii) the last day of our fiscal year following the fifth anniversary of the completion of our
33
IPO on July 25, 2022; (iii) the date on which we have, during the preceding three-year period, issued more than US$1.0 billion in non-convertible debt; or (iv) the date on which we are deemed to be a “large accelerated filer” under the Securities Exchange Act of 1934, as amended, or the Exchange Act, which would occur if the market value of our ordinary shares that are held by non-affiliates exceeds US$700 million as of the last business day of our most recently completed second fiscal quarter. Once we cease to be an emerging growth company, we will not be entitled to the exemptions provided in the JOBS Act discussed above.
Foreign Private Issuer Status
We are incorporated in the Cayman Islands, and (i) the majority of the company’s executive officers or directors are not U.S. citizens or residents; (ii) more than 50% of our assets are located out of the U.S.; and (iii) our business is administered principally out of the U.S. Therefore, we are a “foreign private issuer,” as defined in Rule 405 under the Securities Act and Rule 3b-4(c) under the Exchange Act. As a result, we are not subject to the same requirements as U.S. domestic issuers. Under the Exchange Act, we will be subject to reporting obligations that, to some extent, are more lenient and less frequent than those of U.S. domestic reporting companies. For example, we will not be required to issue quarterly reports or proxy statements. We will not be required to disclose detailed individual executive compensation information. Furthermore, our directors and executive officers will not be required to report equity holdings under Section 16 of the Exchange Act and will not be subject to the insider short-swing profit disclosure and recovery regime. In addition, as a company incorporated in the Cayman Islands, we are permitted to adopt certain home country practices in relation to corporate governance matters that differ significantly from the Nasdaq Stock Market corporate governance requirements. These practices may afford less protection to shareholders than they would enjoy if we complied fully with the Nasdaq Stock Market corporate governance requirements.
B. Business overview.
Virax Biolabs is a holding company incorporated as an exempted company under the laws of the Cayman Islands. As a holding company with no material operations of our own, Virax Biolabs conducts our primary operations through its operating subsidiaries in the United Kingdom, the United States and China and has been operating since 2013.
Virax Biolabs Group Limited and its subsidiaries is a global innovative biotechnology company focused on the prevention, detection, diagnosis, and risk management of viral diseases with a current focus on the field of T cell in Vitro Diagnostics. The Company is in the process of launching an upcoming brand, ViraxImmune which involves developing and manufacturing tests that can predict adaptive immunity to viral diseases as well as identify individuals suffering from T cell exhaustion linked to post viral syndromes. The Company's mission is to protect people from viral diseases and help with the early diagnosis of post viral syndromes associated with T cell exhaustion and chronic fatigue through the provision of diagnostic tests and tests for adaptive immunity.
Initially, we will be focusing on measuring chronic inflammation associated with T cell exhaustion in areas such as SARS-CoV-2 effects, Chimeric Antigen Receptor T cell (CAR-T) therapies, Myloid, Encephalomyelitis (ME) and post viral syndromes. This versatility will position ViraxImmune as a comprehensive platform capable of potentially addressing a wide range of research, diagnostic and health needs within indications associated with chronic inflammation and T cell exhaustion.
The Company's other minor platforms include the ViraxClear and ViraxVet brands, where we may act as a distributor of third-party test kits that have already been approved in the geographic regions we may sell in. Currently, we do not manufacture or develop any product that we sell in our ViraxClear and ViraxVet product portfolios.
Our Products and Services
ViraxClear and ViraxVet
Our existing, minor product portfolio consists of IVD test kits which we have the ability to distribute through our ViraxClear and ViraxVet brands. Currently, we do not manufacture or develop any product that we may sell and we act as a distributor of third-party suppliers’ products. For the fiscal year ended March 31, 2024, revenues generated from these brands were immaterial and accounted for 100% of our total revenues.
The Company has the ability to distribute primarily the following existing test kits under the ViraxClear and ViraxVet brands:
Range |
Test Specification |
Self-Test or Professional |
Respiratory |
SARS CoV-2 Ag |
Both |
FluAB/SARS CoV-2 Ag |
Both |
|
FluAB/SARS CoV-2/RSV Ag |
Both |
|
FluAB/SARS CoV-2/RSV/ADV Ag |
Both |
|
Strep A Ag |
Professional |
34
|
Range of Ag and multiplex PCR based tests for single and multiplex pathogens. Various pack sizes from 1 to 25 units Inc AIV FluAPCR, CoV2 Ab neutralizing, CoV2 Ag saliva, anti-TB, ADV Ag, MycoPneu IgM. |
Professional |
Pregnancy & Fertility |
Rapid and digital pregnancy tests to indicate pregnancy and time from conception |
Both |
LH surge - Ovulation |
Both |
|
FSH - Ovulation |
Both |
|
Sperm concentration tests – male potency |
Both |
|
Infectious Diseases |
Currently sourcing a range of self-test Ag products for STI and other IDs |
Self-Test |
A range of infectious diseases rapid Ag and multiplex PCR based tests. Includes Dengue, HBV, HCV, H Pylorii, GA rota, Noro, Adeno, Typhoid, Malaria, Zika, Chikungunya, EBV quant |
Professional |
|
STI |
A range of STD tests including; HIV (blood and saliva) P24 ag, syphilis, Trichomonas, NG, CT, Candida |
Professional |
Drugs of Abuse |
Multidrug screening panels (inc. saliva) |
Professional |
Oncology |
TraFOB, PSA, Alpha Fetoprotein, carcinoembryonic Ag, |
Professional |
Cardiac |
Inc. Cardiac marker panel, CK, cTnI, |
Professional |
Women’s Health |
Multidrug screening panels incMultiplex PCR assay tests for simultaneous detection of ether 18 or 21 oncogenic HPV genotypes to aid patient management depending on genotype infection. |
Professional |
Other |
Transferrin and CRP tests |
Professional |
ViraxVet |
Various rapid and molecular tests for small animal veterinary diagnostics (Mainly Cats and Dogs) |
Professional |
ViraxImmune
ViraxImmune is our brand under development and also our primary focus in the near future. We are in the process of developing a comprehensive set of T cell diagnostics and immune profiling solutions. Our strategic focus is the development and commercialization of immune profiling IVDs in indications associated with chronic inflammation and T cell exhaustion, such as post viral syndromes and other infectious diseases, chronic inflammation and immune-oncology.
The potential applications of ViraxImmune are not restricted to any single disease or condition. The platform's capabilities will initially focus on measuring chronic inflammation associated with T cell exhaustion in areas such as SARS-CoV-2 effects, Chimeric Antigen Receptor T cell (CAR-T) therapies, Myloid, Encephalomyelitis (ME) and post viral syndromes. This versatility will position ViraxImmune as a comprehensive platform capable of potentially addressing a wide range of research, diagnostic and health needs within indications associated with chronic inflammation and T cell exhaustion.
Post-viral syndromes are a wide range of complex conditions involving physical, cognitive, emotional and neurological difficulties that vary in severity over time. These conditions frequently lead to a sense of tiredness and weakness, pain, difficulty concentrating and headaches that linger after the viral infection has cleared. Symptoms may continue for weeks, months or longer. Various microbial and viral infections are possible trigger factors for post-viral syndrome, including SARS-CoV-2, Epstein-Barr virus, cytomegalovirus, human herpesvirus, enteroviruses, HPV, Lyme disease and Zika, among others.
The diagnosis of these conditions can be very difficult among sufferers who are often neglected by healthcare systems that are unable to identify a definitive diagnosis and treatment strategy. The first point of diagnosis should be identifying that there is an issue and pinpointing the initial cause. The ViraxImmune diagnostic test may help to identify if an individual is suffering from a post-viral syndrome and also elucidate the original viral cause of the condition. The ViraxImmune diagnostic may also identify elements associated with these conditions, such as a persistent cytokine response in addition to T cell exhaustion.
As an example Long COVID represents a significant public health challenge in the post-SARS-CoV-2 era. Epidemiological studies indicate that 10-30% of individuals infected with SARS-CoV-2 experience persistent symptoms beyond the acute phase of the infection. These symptoms, including chronic fatigue, cognitive dysfunction, and respiratory difficulties, persistently affect patient health and quality of life. The heterogeneity of these symptoms complicates the diagnostic process, as they overlap with a range of other post-viral syndromes, posing a challenge to healthcare professionals. The diagnostic tools currently available such as primarily PCR and lateral flow tests, are optimized for acute viral infection detection and do not adequately address the chronic and complex nature of Long COVID. Consequently, many patients experience a diagnostic ambiguity, impacting their subsequent treatment and care. This challenge is exacerbated by the high prevalence of SARS-CoV-2 exposure, both through infection and vaccination, necessitating the development of diagnostic methods specifically tailored to Long COVID.
The primary markets of interest will be the United Kingdom and North America due to a rise in chronic infections, rising healthcare expenditures, a well-developed healthcare sector, large patient population, and government support for research and development. Virax Biolabs will also look to gain regulatory approval of the IVDs in these territories followed by the European Union. The key revenue
35
streams are anticipated to be healthcare providers treating patients, and researchers and drug companies developing treatments in the indications of interest.
The Company intends to initially launch a number of T cell immune profiling assays in different indications as research-use-only ("RUO") followed by the later release of IVDs centered around T cell exhaustion and chronic inflammation through the ViraxImmune platform. The development of a fully approved IVD will involve performing the appropriate analytical and clinical performance studies required as part of the regulatory requirements in the different territories. As part of the development of the in-vitro diagnostic ("IVD") the Company intends to work alongside the appropriate governmental healthcare providers to allow the test to be incorporated into the healthcare system and pharmacy outlets. All intellectual property rights developed during the course of the research and development activities will belong to the Company. To guarantee the relevance of our clinical evidence, the study protocol will be discussed with the FDA and MHRA (UK) via a pre-submission process for guidance prior to the start of any clinical performance study.
Our Industry
Our Company competes in the IVD market. The IVD tests are defined as medical devices and reagents that are used to analyze specimens derived from the human body (including blood, tissues, and other body fluids) to detect diseases, conditions, and infections. IVD tests are usually performed at either stand-alone laboratory, hospital-based laboratory, or point-of-care (“POC”) centers. The technologies used for test sample preparation majorly include polymerase chain reaction (“PCR”), microarray techniques, sequencing technology, and mass spectrometry. Based on the key technologies involved, the global IVD market is fragmented into sub-segments including Immunoassay, Hematology, Clinical Chemistry, Molecular Diagnostics, Microbiology, Haemostasias, Flow Cytometry and others.
The IVD sector is expected to experience growth rates owing to driving factors such as aging global population, increase in the occurrence of complex infectious diseases, an increase in awareness among the global urban populations, etc. However, lack of proper reimbursement policies in the developing nations and skepticism among patients to get regular healthcare consulting are still hindrances in some regions, especially third-world countries, which impedes the growth of the IVD market.
Key Supplier Relationships
We source our suppliers through multiple channels through referrals from counterparties and through industry exhibitions/expos. Our suppliers are divided into two categories: (1) those manufacturing our products as per our manufacturing standards, and (2) those providing products for our distribution. Our Company works closely with our suppliers to assure continuity of supply while maintaining high quality and reliability. To date, our Company has not experienced any significant difficulty in locating and obtaining the materials necessary to fulfill our production schedules.
Key Customer Relationships
Our Company has two types of customers: (i) direct end user customers, which may include research organizations, corporations, independent laboratories, large hospital systems and public and private institutions, and (ii) distributor customers, which distribute our own brands and products we sourced from third party suppliers.
Our Competitive Strengths
We believe the following competitive strengths differentiate us from our competitors and will continue to contribute to our success:
Cutting-edge technology
Our Company is engaged in creating a cutting-edge technology. In particular, our in-development ViraxImmune’s immunological diagnostic profiling technique is intended to be cutting-edge technology which we believe is not currently available on the IVD market as at the date of this report. Currently, we are in the process of developing a comprehensive set of T cell diagnostics and immune profiling solutions. Our strategic focus is the development and commercialization of immune profiling IVDs in indications associated with chronic inflammation and T cell exhaustion, such as post viral syndromes and other infectious diseases, chronic inflammation and immune-oncology.
The potential applications of ViraxImmune are not restricted to any single disease or condition. The platform's capabilities will initially focus on measuring chronic inflammation associated with T cell exhaustion in areas such as SARS-CoV-2 effects, Chimeric Antigen Receptor T cell (CAR-T) therapies, Myloid, Encephalomyelitis (ME) and post viral syndromes. This versatility will position ViraxImmune as a comprehensive platform capable of potentially addressing a wide range of research, diagnostic and health needs within indications associated with chronic inflammation and T cell exhaustion.
36
Commercialization of our own diagnostic devices
Historically in-vitro diagnostic test kits are designed to be lab specific by leading biotechnology and pharmaceutical companies, and thus, an in-vitro diagnostic test kit company is required to be tied down to a specific biotechnology partner or pharmaceutical partner. However, we are designing our ViraxImmune T cell IVD/Immune response Test kit to be as lab agnostic and easy to use as possible. As a result, we believe this will allow us to distribute the T cell in-vitro diagnostics test kit to a broader geographic reach and deploy the test kits rapidly, without having to impose difficult techniques or equipment on our lab partners or being tied down to a specific lab partner. As a result, we believe we can rapidly capture the T cell in-vitro diagnostics test kit market share in a short period of time.
We are concentrating on developing diagnostics for the early diagnosis for post viral syndromes and indications associated with chronic inflammation and T cell exhaustion as there is a major medical need for early detection to enable early treatment interventions. The primary markets of interest will be the United Kingdom and North America due to a rise in chronic infection, rising healthcare expenditures, a well-developed healthcare sector, rising healthcare expenditures, large patient population, and government support for research and development.
Experienced Management Team with Extensive Industry Expertise and a Global Vision
Our Company has an experienced management team driven by a shared passion for the prevention, detection, diagnosis and risk management of viral diseases, particularly immunology. Our Company is led by our chief executive officer, Mr. James Foster, who had entrepreneurial successes in various investment companies before co-founding Virax. In 2009, Mr. Foster co-founded Cryptex Card, one of the world's first global debit card companies for Bitcoin. In 2013, Mr. Foster co-founded Natural Source Group Pte. Limited, a company which received venture capital funding. Our Chief Operating Officer is Dr. Nigel McCracken. Prior to joining Virax, Dr. McCracken was the Chief Scientific Officer at OTC traded BerGenBio AsA where he drove the plan for BerGenBio AsA's companion diagnostics and assay development. Prior to that role, he served as COO at Nasdaq listed NuCana PLC, a clinical-stage biopharmaceutical company focused on significantly improving treatment outcomes for patients with cancer, where he provided oversight and direction to the operations of the business, research and development, and execution of corporate strategy. Dr. McCracken obtained a Master's degree in Clinical Pharmacology from Kings College London, a Doctor of Philosophy degree in Biochemical Toxicology from the University of Newcastle Upon Tyne, and a Bachelor of Science degree in Biochemistry and Pharmacology from Strathclyde University. Other members of our management team are also industry veterans with diverse expertise, such as in developing advanced technology platforms, as well as overseeing investments, financing and other corporate development initiatives of various companies, and possess keen insights into the latest trends in the global healthcare and pharmaceutical market.
Expanding Research and Development Capabilities
Our Company has and is continuing to invest significant resources in research and development. For the years ended March 31, 2024 and 2023, our research and development expenses were $1,561,965 and $2,179,341, respectively. As of March 31, 2024, we have an intellectual property portfolio consisting of 2 pending patents, 3 pending trademarks and 4 registered domain names. We have built a strong research and development team and are developing a T cell IVD/Immune response Test kit under the ViraxImmune brand and will initially focus on measuring chronic inflammation associated with T cell exhaustion in areas such as SARS-CoV-2 effects, Chimeric Antigen Receptor T cell (CAR-T) therapies, Myloid, Encephalomyelitis (ME) and post viral syndromes. We will subsequently be addressing a wide range of research, diagnostic and health needs within indications associated with chronic inflammation and T cell exhaustion. As of March 31, 2024, our research and development team was composed of 9 personnel, which accounted for approximately 53% of our total employees. Our research and development team has years of technology know-how in developing and launching products and services in response to market demands. We believe this can lead to a shorter time to market which in turn may allow us to fully capture opportunities presented by shifts in industry trends. Further, our in-house research and development team collaborate closely with our manufacturing and research and development partners to ensure our products receive timely updates and/or the new biotechnology to keep abreast of global outbreaks of viral diseases.
Our Strategies
Development of the proprietary ViraxImmune suite of IVD T cell test kits.
Post-viral syndromes are a wide range of complex conditions involving physical, cognitive, emotional and neurological difficulties that vary in severity over time. These conditions frequently lead to a sense of tiredness and weakness, pain, difficulty concentrating and headaches that linger after the viral infection has cleared. Symptoms may continue for weeks, months or longer. Various microbial and viral infections are possible trigger factors for post-viral syndrome, including SARS-CoV-2, Epstein-Barr virus, cytomegalovirus, human herpesvirus, enteroviruses, HPV, Lyme disease and Zika, among others.
The diagnosis of these conditions can be very difficult among sufferers who are often neglected by healthcare systems that are unable to identify a definitive diagnosis and treatment strategy. The first point of diagnosis should be identifying that there is an issue and pinpointing the initial cause. The ViraxImmune diagnostic test may help to identify if an individual is suffering from a post-viral
37
syndrome and also elucidate the original viral cause of the condition. The ViraxImmune diagnostic may also identify elements associated with these conditions, such as a persistent cytokine response in addition to T cell exhaustion. For further details on ViraxImmune, see “Our Products and Services — ViraxImmune” in this section.
Launch Expanded Sales and Marketing.
We intend to strengthen and expand our sales and marketing efforts by utilizing the following strategies, among others:
Sales, Distribution, Marketing and Advertising
Our Company is continuing to build sales and distribution networks.
Our marketing strategy will largely focus on educating consumers, in particular corporate consumers, about our products as everyone may potentially be susceptible to a viral disease. We also plan to focus on clinics, pharmacies, laboratories, hospitals, and other relevant groups once we receive regulatory approval for our ViraxImmune product. We will use a combination of techniques in our marketing approach including but not limited to social media campaigns and public relations.
Research and Development
For years ended March 31, 2024 and 2023, our research and development expenses amounted to $1,561,965 and $2,179,341, respectively. As of March 31, 2024, our internal research and development team was composed of 9 personnel, who have expertise in developing and manufacturing diagnostics as well as expertise in immunology and within infectious disease and oncology.
Our research and development strategy has been centered on:
Intellectual Property
Our success and future revenue growth depend, in part, on our ability to protect our intellectual property. We rely primarily on patent, copyright, trademark and trade secret laws, as well as confidentiality procedures, to protect our proprietary technologies and processes.
We believe that the core of our business is comprised of our proprietary technologies. As a result, we strive to maintain a robust intellectual property portfolio. Our success and future revenue growth may depend, in part, on our ability to protect our intellectual property as products and services that are material to our operating results incorporate patented technology.
38
We have pursued rights in intellectual property, and we focus our intellectual property efforts globally. Our patent strategy is designed to provide a balance between the need for coverage in our strategic market and the need to maintain reasonable costs.
We believe our rights to patents, copyrights, trademarks and other intellectual property rights serve to distinguish and protect our products from infringement and contribute to our competitive advantages.
As of the date of this report, we have applied for 2 patents, including exemplary jurisdictions where patent applications have been filed, and expected expiration dates are summarized in the following table:
NO. |
|
ITEM |
|
JURISDICTIONS |
|
PATENT/ |
|
EXPIRATION* |
|
TYPE |
1. |
|
Methods of detecting T Cells |
|
Global |
|
GB 2201765.1 Pending |
|
February 2043 |
|
Utility |
2. |
|
Peptide Pools derived from Viruses |
|
Global |
|
GB 2201768.5 Pending |
|
February 2043 |
|
Utility |
* The expiration dates assume that non-provisional patent applications will be filed approximately one year after the earliest priority date and that national stage applications will be filed, as appropriate, and pursued until grant, and that all renewal and annuity fees will be paid.
In most countries worldwide, the term of a utility patent expires 20 years from the earliest effective non-provisional filing date, subject to the timely payment of the requisite annuities or other renewal fees.
We cannot assure you that any pending patent or copyright will be approved by the relevant government authorities. In addition, any rights granted under any of our existing or future patents, copyrights or trademarks may not provide meaningful protection or any commercial advantage to us. With respect to our other proprietary rights, it may be possible for third parties to copy or otherwise obtain and use proprietary technology without authorization or to develop similar technology independently. We may in the future initiate claims or litigation against third parties to determine the validity and scope of proprietary rights of others. In addition, we may in the future initiate litigation to enforce our intellectual property rights or to protect our trade secrets. Additional information about the risks relating to our intellectual property is provided under “Risk Factors — Risks Related to Intellectual Property.”
Competition
We face significant competition in our evolving industries from numerous competitors, particularly the in-vitro diagnostics industry. To differentiate us from other in-vitro diagnostics providers in the industry, we focus on cutting-edge technology capable of potentially addressing a wide range of research, diagnostic and health needs within indications associated with chronic inflammation and T-cell exhaustion.
Participants in the in-vitro diagnostics industry include biotechnology companies, established pharmaceutical companies, and other in-vitro diagnostics companies. Many of our competitors developed in-vitro diagnostic test kits and other products similar to us. As of the date of this report, we consider our main IVD competitors to be Qiagen N.V. (NYSE: QGEN), Adaptive Biotechnologies Corporation (NASDAQ: ADPT), Roche Holding AG (SIX: ROG) and Abbott Laboratories (NYSE: ABT). We may also face competition from new and emerging companies.
Compared to our company, our current and potential competitors may have:
However, we believe we are well positioned to compete in the in-vitro diagnostics market as a result of our comprehensive product portfolio, research and development capabilities, and experienced management team.
The principal competitive factors in the in-vitro diagnostics market include:
39
We believe we compete favorably with respect to the factors mentioned above.
Regulations
This section sets forth a summary of the significant regulations or requirements in the jurisdictions where we conduct our material business operations, namely the United Kingdom. The primary Singapore laws and regulations, which do not purport to be complete, to which we are subject relate to foreign investment, dividend distributions, foreign exchange controls, data protection, intellectual property rights, anti-money laundering and terrorism financing and employment and labor. This section also sets forth a summary of regulatory approval on medical device products for the relevant jurisdictions for IVD and a summary of the relevant PRC laws, regulations and government policies that are relevant to Shanghai Xitu in the PRC.
United Kingdom
The UK’s withdrawal from the EU will have major ramifications for IVD manufacturers that will, among other things have to follow new procedures that will apply in the UK including appointment of a UK Responsible Person rather than relying on European Authorized Representatives to manage their compliance efforts in the UK.
IVDs compliant with the EU in vitro diagnostic medical devices regulation (EU IVDD) can be placed on the Great Britain market up until the 30 June 2030. We intend to use the recognized CE marks that we will apply with the European Union for our medical device product, namely our current in development T cell IVD Test under the ViraxImmune brand. After which, we will apply with the UK Medicine and Healthcare Products Regulatory Agency for a UK Conformity Assessed mark.
United States
The FDA regulates the sale or distribution of medical devices, including but not limited to IVD test kit. IVD products are subject to regulations by the FDA as medical devices to the extent that they are intended for use in the diagnosis, treatment, cure, mitigation or prevention of disease or other conditions.
The information that must be submitted to the FDA in order to obtain clearance or approval to market a new medical device varies depending on how the medical device is classified by the FDA. Medical devices are classified into one of three risk-based classes depending on the controls deemed by the FDA to be necessary to reasonably ensure their safety and effectiveness. Class I devices are considered the lowest risk and are subject to general controls, including labeling requirements, and adherence to the FDA’s quality system regulations, or QSRs, which are device-specific current good manufacturing practices. Class II (intermediate risk) devices may be subject to premarket notification, or may be 510(k)-exempt, and are generally subject to QSRs, general controls and sometimes special controls, including performance standards and post-market surveillance. Class III (highest risk) devices are subject to most of the previously identified requirements as well as to pre-market approval. Class I devices are exempt from premarket review; most Class II devices require 510(k) clearance, and all Class III devices must receive premarket approval before they can be sold in the United States. The payment of a user fee, which is typically adjusted annually, to the FDA is usually required when a 510(k) notice or premarket approval application is submitted.
510(k) Premarket Notification
A 510(k) premarket notification requires the sponsor to demonstrate that a medical device is substantially equivalent to another marketed device, termed a “predicate device,” that is legally marketed in the United States and for which a premarket approval was not required. A device is substantially equivalent to a predicate device if it has the same intended use and technological characteristics as the predicate; or has the same intended use but different technological characteristics, where the information submitted to the FDA does not raise new questions of safety and effectiveness and demonstrates that the device is at least as safe and effective as the legally marketed device.
If the FDA believes that the device is not substantially equivalent to a predicate device, it will issue a “Not Substantially Equivalent” (“NSE”) determination and designate the device as a Class III device, which will require the submission and approval of a PMA before the new device may be marketed. A person who receives an NSE determination in response to a 510(k) submission may, within 30 days of receipt of the NSE determination, submit a de novo request for the FDA to make a risk-based evaluation for classification of the device into Class I or II. Devices that are classified through the de novo process may be marketed and used as predicates for future 510(k) submissions. The FDA continues to reevaluate the 510(k) pathway and process and the de novo process, and has taken what it
40
describes as a risk-based approach to develop innovative regulatory policy to propose a more “contemporary” approach. In October 2017, the FDA published a final guidance entitled, “De Novo Classification Process (Evaluation of Automatic Class III Designation),” and in December 2018, the FDA published a proposed rule which if finalized is intended to provide structure, clarity and transparency on the de novo classification process. In January 2021, it also published a final guidance entitled “Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program.”
Pre-market Approval (“PMA”)
A PMA must be supported by more detailed and comprehensive scientific evidence, including clinical data, to demonstrate the safety and efficacy of the medical device for its intended purpose. If the device is determined to present a “significant risk,” the sponsor may not begin a clinical trial until it submits an investigational device exemption (“IDE”) to the FDA and obtains approval to begin the trial.
After a PMA is submitted, the FDA has 45 days to make a threshold determination that the PMA is sufficiently complete to permit a substantive review. If the PMA is complete, the FDA will file the PMA. The FDA is subject to a performance goal review time for a PMA that is 180 days from the date of filing, although in practice this review time is longer. Questions from the FDA, requests for additional data and referrals to advisory committees may delay the process considerably. The total process may take several years and there is no guarantee that the PMA will ever be approved. Even if approved, the FDA may limit the indications for which the device may be marketed. The FDA may also request additional clinical data as a condition of approval or after the PMA is approved. Any changes to the medical device may require a supplemental PMA to be submitted and approved before changed medical device may be marketed.
Any products sold by us pursuant to FDA clearances or approvals will be subject to pervasive and continuing regulation by the FDA, including record keeping requirements, reporting of adverse experiences with the use of the device and restrictions on the advertising and promotion of our products. In particular, we may not advertise or otherwise promote our devices for indications, patient populations, or other conditions of use that are not covered by the applicable FDA clearance or approval for the device. Modifications or changes to the device or how it is manufactured may also be separately subject to FDA review and authorization before being commercialized. Device manufacturers are required to register their establishments and list their devices with the FDA and are subject to periodic inspections by the FDA and certain state agencies. Noncompliance with applicable FDA requirements can result in, among other things, warning letters, fines, injunctions, civil penalties, recalls or seizures of products, total or partial suspension of production, refusal of the FDA to grant 510(k) clearance or PMA approval for new devices, withdrawal of 510(k) clearances and/or PMA approvals and criminal prosecution.
As a result of the SARS-CoV-2 pandemic, the US government declared a state of emergency which enabled the FDA to issue emergency use authorizations (“EUAs”) to provide more timely access to critical medical products (including medicines and tests) that may help during the emergency when there are no adequate, approved, and available alternative options. EUAs are in effect until the emergency declaration ends but can be revised or revoked as FDA considers the needs during the emergency and new data on a product’s safety and effectiveness, or as products meet the criteria to become approved, cleared, or licensed by the FDA. Manufacturers of several types of SARS-CoV-2 assays have been granted EUAs. These authorizations are only intended for the duration of the emergency declaration, and afterwards will be revoked. The FDA has indicated the withdrawal of the EUA process will be done in a controlled ramp down.
Additionally, we may be required to conduct costly post-market testing, and we will be required to report adverse events and malfunctions related to our products. Later discovery of previously unknown problems with our products, including unanticipated adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements may result in restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recalls, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties.
Furthermore, the relevant authorities will inspect our facilities and those of our suppliers from time to time to determine whether we are in compliance with regulations relating to the manufacture of diagnostic products, including regulations concerning design, manufacture, testing, quality control, product labeling, distribution, promotion and record-keeping practices. A determination that we are in material violation of such regulations could lead to the imposition of civil penalties, including fines, product recalls, product seizures or, in extreme cases, criminal sanctions.
PRC laws and regulations applicable to Shanghai Xitu
As illustrated in “Corporate History and Structure” Shanghai Xitu Consulting Co., Limited (“Shanghai Xitu”) is our Company's sole PRC subsidiary. Shanghai Xitu is a wholly owned subsidiary of Logico BVI, and is classified as a wholly foreign owned enterprise (“WFOE”) under PRC law.
Regulations Related to Business Registration
According to the Foreign Investment Law of China released by The National People’s Congress of the People’s Republic of China on March 15, 2019, to set up a WFOE in China:
41
The Special Administrative Measures (Negative List) for Foreign Investment Access (Edition 2021) released by the National Development and Reform Commission (“NDRC”) and MOFCOM on 27 December 2021 have set out the industries in which foreign investment is prohibited or restricted. If a PRC company engages in business in certain industries (such as the finance industry), it may need to obtain special license or approval from the relevant authority in addition to its business license.
Each PRC company has a “business scope” set out on its business license. The PRC company may conduct business within such scope. Further, according to the current PRC law and legal practice, a company may also conduct activities outside of its registered business scope unless any special license/approval is required for such additional business activities.
Upon management's review, Shanghai Xitu has obtained the Foreign Investment Approval Certificate issued by the MOFCOM. The registered business scope of Shanghai Xitu as shown on its business license is as follows: “business information consultation, business management consultation, business registration agency, marketing planning, corporate image planning, conference services (except for hosting, undertaking, and exhibitions), exhibition services (hosting, undertaking, excluding exhibitions), technology development in the field of network technology, proprietary technology transfer, technology consulting, and technical services.”
Upon management's review of the Negative List and Business Scope Specification Expression Query System, as at the date of this report, the registered business scope of Shanghai Xitu is not on the Negative List. As a result, there is no prohibition or restriction on foreign investment in such industries.
In addition, Shanghai Xitu does not need to obtain any special license or approval granted by the relevant authority for it to conduct business within its registered business scope. The business license of Shanghai Xitu is sufficient for it to conduct its registered business scope. Upon management's review, Shanghai Xitu is primarily engaged in procurement. Currently, it procures the relevant medical goods from China for its affiliate in Singapore by signing contracts with the PRC suppliers on behalf of its said affiliate.
Shanghai Xitu’s current business is not stated on its registered business scope. As stated at above, a PRC company may conduct activities outside of its registered business scope unless any special license/approval is required for such additional business activities. For such current business of Shanghai Xitu, it is not required to obtain any special license/approval. Therefore, Shanghai Xitu may conduct such activities beyond its registered business scope.
C. Organizational structure.
Our corporate structure consists of Virax Biolabs Group Limited and our wholly owned subsidiaries, described below.
Virax Biolabs Group Limited (“Virax Biolabs”) — Virax Biolabs Group Limited is a Cayman Islands exempted company incorporated with limited liability on September 2, 2021.
Virax Biolabs (UK) Limited (“Virax UK”) — Virax Biolabs (UK) Limited was incorporated on August 19, 2021 under the laws of the United Kingdom, a wholly-owned subsidiary of the Company and primarily engaged in the Company's research and development activities.
Virax Biolabs Limited (“HKCo” or formerly known as Shanghai Biotechnology Devices Ltd.) — Virax Biolabs Limited, incorporated on April 14, 2020, under the laws of Hong Kong, was previously named as “Shanghai Biotechnology Devices Limited” and effected a name change to “Virax Biolabs Limited” on July 12, 2021. Virax Biolabs Limited, our wholly owned Hong Kong subsidiary, serves as a holding company.
Virax Immune T- Cell Medical Device Company Limited (“Virax Immune T cell”) — Virax Immune T cell Medical Device Company Limited, a wholly-owned subsidiary of HKCo, incorporated on January 16, 2017, under the laws of Hong Kong, was previously named as “Stork Nutrition Asia Limited” and effected a name change to “Virax Immune T cell Medical Device Company Limited” on September 10, 2021.
Virax Biolabs Pte. Limited (“SingaporeCo”) — Virax Biolabs Pte. Limited, incorporated on May 4, 2013 under the laws of Singapore, was previously named as “Natural Source Group Pte. Limited” and effected a name change to “Virax Biolabs Pte. Limited” on July 2, 2021. 95.54% of its capital stock is owned by Virax Biolabs Limited and the remaining 4.35% by independent third-party shareholders.
42
Logico Bioproduct Corp. (“Logico BVI”) — Logico Bioproducts Corp., a wholly-owned subsidiary of SingaporeCo, is a limited liability company incorporated in the British Virgin Islands on January 21, 2011 and is a holding company.
Shanghai Xitu Consulting Co., Limited (“Shanghai Xitu”) — Shanghai Xitu, a wholly-owned subsidiary of Logico BVI and a wholly foreign owned enterprise, is a limited liability company incorporated on October 27, 2017, in China. Shanghai Xitu is primarily engaged in procurement, warehousing, product development, and staffing management.
Virax Biolabs USA Management, Inc. — Virax Biolabs USA Management, Inc. was incorporated on August 1, 2022 under the laws of the United States, a wholly-owned subsidiary of Virax Biolabs and structured as a management company for operations within the United States.
Virax Biolabs Group Holdings Ltd (“Virax UK HoldCo”) — Virax Biolabs Group Holdings Limited was incorporated on February 22, 2023 under the laws of the United Kingdom, a wholly-owned subsidiary of the Company and structured as a holding company.
Virax Biolabs FZ-LLC (“Virax Dubai”) — Virax Biolabs FZ-LLC was incorporated on April 18, 2023 under the laws of the United Arab Emirates, a wholly-owned subsidiary of the Company and is primarily engaged as a regional distribution company.
Virax Biolabs Trading B.V. (“Virax Netherlands”) — Virax Biolabs Trading B.V. was incorporated on August 4, 2023 under the laws of the Netherlands, a wholly-owned subsidiary of the Company and is primarily engaged as a regional distribution company.
Virax Biolabs UK Operating Limited (“Virax UK Operating”) — Virax Biolabs UK Operating Limited was incorporated on November 7, 2023 under the laws of the United Kingdom, a wholly-owned subsidiary of the Company and is primarily engaged as a regional operating company.
The following diagram illustrates our corporate structure:
D. Property, plant and equipment.
43
We are headquartered in Glasgow, United Kingdom. In Glasgow, we lease lab space which consists of approximately 198 square meters of space. Lease payments are approximately GBP 3,900 per month. This lab space houses our research operations and has the capacity to fully support any future production of our anticipated products. We also lease lab space in London which consists of approximately 300 square meters of space. Lease payments are approximately GBP 9,600, per month. This lab space lease expires in June 2024 and will not be renewed. In Shanghai, we lease office space which consists of approximately 99 square meters of space. Lease payments are approximately RMB 21,600, per month. This office space houses our China procurement team and will not be renewed.
The majority of our tangible property, plant and equipment consists of laboratory equipment which is fully capable of handling all of the Company's research and development needs as well as future production needs. As of March 31, 2024, the tangible net book value of our property, plant and equipment is $897,627.
Item 4A. Unresolved Staff Comments
Not applicable.
Item 5. Operating and Financial Review and Prospects
The following discussion and analysis of our financial condition and results of operations for the fiscal years ended March 30, 2024 and 2023 should be read in conjunction with our consolidated financial statements and the related notes included elsewhere in this annual report. Our consolidated financial statements have been prepared in accordance with IFRS. Some of the information contained in this discussion and analysis or set forth elsewhere in this annual report, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. See “Forward-Looking Statements” for a discussion of the uncertainties, risks and assumptions associated with these statements. Actual results and the timing of events could differ materially from those discussed in our forward-looking statements as a result of many factors, including those set forth under “Risk Factors” and elsewhere in this annual report.
A. Operating results.
Overview
Virax Biolabs is a holding company incorporated as an exempted company under the laws of the Cayman Islands. As a holding company with no material operations of its own, it conducts the majority of its operations through its subsidiaries in the United Kingdom, the United States, and China and has been operating since 2013.
The Company is a global innovative biotechnology company focused on the prevention, detection, diagnosis, and risk management of viral diseases with a current focus on the field of T cell in Vitro Diagnostics. The Company is in the process of developing and manufacturing tests that can predict adaptive immunity to viral diseases as well as identify individuals suffering from T cell exhaustion and chronic inflammation linked to post viral syndromes. The Company's mission is to protect people from viral diseases and help with the early diagnosis of post viral syndromes associated with T cell exhaustion and chronic inflammation through the provision of diagnostic tests and tests for adaptive immunity.
The Company is in the process of developing ViraxImmune, with the intention of providing an immunology profiling platform that assesses each individual’s immune risk profile against major global viral diseases as well as helping with the early diagnosis of post viral syndromes associated with T cell exhaustion and chronic inflammation. We are in the process of developing a T cell Test under the ViraxImmune brand and will apply for regulatory agency approval in the future. We believe that the T cell Tests and immunology platform we are developing under the ViraxImmune brand will be particularly useful in assisting in the threat analysis of the major viruses faced globally as well as helping with the early diagnosis of indications associated with T cell exhaustion and chronic inflammation which have been linked to symptoms such as chronic fatigue. Initially, we will be focusing in diseases associated with post viral syndromes including but not limited to SARS-CoV-2, Human Papillomavirus (better known as HPV), Malaria, Hepatitis B, and Herpes (better known as HSV-1).
The Company also has the ability to distribute existing diagnostics test kits through our ViraxClear and ViraxVet brands. Currently, we do not manufacture or develop any product that we sell in our ViraxClear and ViraxVet product portfolios, and we act as a distributor of third-party suppliers’ products. While ViraxClear and ViraxVet have the ability to sell and distribute existing test kits, they are not the main focus of the Company.
Share Consolidation
The Company's board of directors and shareholders approved a 1-for-10 share consolidation (the “Share Consolidation”) on November 3, 2023 and December 6, 2023, respectively. The Company filed the third amended and restated articles of association (the “Post-Split Amended and Restated Articles of Association”), which became effective on December 18, 2023. Beginning with the opening of trading
44
on December 18, 2023, the Company's ordinary shares began trading on a post-Share Consolidation basis on the Nasdaq Capital Market under the same symbol "VRAX", but under a new CUSIP number of G9495L125. The objective of the Share Consolidation was to enable the Company to regain compliance with Nasdaq Marketplace Rule 5550(a)(2) and maintain its listing on the Nasdaq Capital Market. On January 4, 2024, the Company received notification that it had regained compliance with Nasdaq Marketplace Rule 5550(a)(2).
Upon the effectiveness of the Share Consolidation, every ten issued and outstanding ordinary shares of a par value of US$0.0001 each was automatically consolidated into one issued and outstanding ordinary share of a par value of US$0.001 each. No fractional shares were issued as a result of the Share Consolidation. Instead, any fractional shares that would have resulted from the Share Consolidation were rounded up to the next whole number. The Share Consolidation affected all shareholders uniformly and did not alter any shareholder's percentage interest in the Company's outstanding ordinary shares, except for adjustments that may result from the treatment of fractional shares. As such, all share and per share amounts have been given retroactive effect in the financial statements for all periods presented.
Results from Operations
The following table shows selected audited consolidated statement of profit or loss data for the years ended March 31, 2024 and 2023. For a discussion of our results of operations for the year ended March 31, 2022, including a year-to-year comparison between the years ended March 31, 2023 and 2022, refer to Part I, Item 5, “Operating and Financial Review and Prospects” in our Annual Report Form 20-F for the year ended March 31, 2023.
Years Ended March 31, 2024 and 2023
|
|
For the Year Ended March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Revenues |
|
$ |
156,419 |
|
|
$ |
8,561 |
|
Cost of revenues |
|
|
105,829 |
|
|
|
9,926 |
|
Gross profit (loss) |
|
|
50,590 |
|
|
|
(1,365 |
) |
|
|
|
|
|
|
|
||
Operating Expenses |
|
|
|
|
|
|
||
General and administrative |
|
$ |
4,594,743 |
|
|
$ |
3,552,055 |
|
Research & Development |
|
|
1,561,965 |
|
|
|
2,179,341 |
|
Impairment of intangible asset |
|
|
390,355 |
|
|
|
— |
|
Total operating expenses |
|
|
6,547,063 |
|
|
|
5,731,396 |
|
|
|
|
|
|
|
|
||
Operating loss |
|
$ |
(6,496,473 |
) |
|
$ |
(5,732,761 |
) |
|
|
|
|
|
|
|
||
Other income (expense), net |
|
|
(242,647 |
) |
|
|
274,998 |
|
|
|
|
|
|
|
|
||
Net loss |
|
$ |
(6,739,120 |
) |
|
$ |
(5,457,763 |
) |
|
|
|
|
|
|
|
||
Other comprehensive loss |
|
|
|
|
|
|
||
Foreign currency adjustment |
|
|
(3,639 |
) |
|
|
(111 |
) |
Total Comprehensive Loss |
|
$ |
(6,735,481 |
) |
|
$ |
(5,457,652 |
) |
Revenues
Revenues were $156,419 for the year ended March 31, 2024 which consisted of sales of test kits to two customers. Revenues were $8,561 for the year ended March 31, 2023 which consisted of sales of test kits to one customer.
Cost of revenues
Cost of revenue for the years ended March 31, 2024 and 2023 was $105,829 and $9,926 and which consisted of test kit supply cost from the manufacturer associated with the sales from our ViraxClear test kit distribution discussed above.
Operating Expenses
Operating expenses were $6,547,063 and $5,731,396 for the years ended March 31, 2024 and 2023, respectively, representing an increase of approximately 14%.
45
|
|
For the Year Ended March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Operating expenses: |
|
|
|
|
|
|
||
General and administrative |
|
$ |
4,594,743 |
|
|
$ |
3,552,055 |
|
Research and Development |
|
|
1,561,965 |
|
|
|
2,179,341 |
|
Impairment of intangible asset |
|
|
390,355 |
|
|
|
— |
|
Total operating expenses |
|
$ |
6,547,063 |
|
|
$ |
5,731,396 |
|
Components of Operating Expenses are as follows:
General and Administration - For the years ended March 31, 2024 and 2023, general and administrative costs were $4,594,743 and $3,552,055, respectively. General and administrative personnel costs for the year ended March 31, 2024 was $1,177,001, an increase of $236,416 from $940,585 for the year ended March 31, 2023. Information technology costs for the year ended March 31, 2024 was $138,010, an increase of $118,629 from $19,381 for the year ended March 31, 2023. The new Glasgow lab rental costs for the year ended March 31, 2024 was $193,571, an increase of $163,005 from $30,566 for the year ended March 31, 2023. Depreciation and amortization for the year ended March 31, 2024 was $102,932, while there was no depreciation or amortization expense for the year ended March 31, 2023. Directors and officers insurance for the year ended March 31, 2024 was $568,175, an increase of $78,021 from $490,154 for the year ended March 31, 2023. These increases were offset by a decrease in stock-based compensation. Stock-based compensation allocated to general and administrative costs for the year ended March 31, 2024 was $943,876, an increase of $262,760 from $943,876 for the year ended March 31, 2023.
Research and Development - For the year ended March 31, 2024, research and development expenses consisted of $982,339 in payroll and stock-based compensation costs and $579,626 in materials and consumables associated with the ongoing clinical studies for the ViraxImmune platform. For the year ended March 31, 2023, research and development expenses consisted of $448,031 in clinical protocol and performance studies from third party laboratory partners as well as $1,731,310 in payroll and stock-based compensation costs.
Impairment of intangible assets - For the year ended March 31, 2024, there was an impairment charge of $390,355 for the ViraxImmune Mobile Application, which development has been suspended and has not been placed into services as of the date of this report. Based on the estimated projections of the future cashflows of the application, the Company concluded that since the ViraxImmune mobile application has been suspended, and the present value of future cashflows does not exceed the carrying value of the capitalized amount, there is a full impairment of these amounts at March 31, 2024. There was no impairment during the year ended March 31, 2023.
Income tax expense
There was no income tax expense for the years ended March 31, 2024 and 2023.
Total other Income, Expense, net
For the year ended March 31, 2024, our total other net expenses were $242,647. For the ended March 31, 2023, our total other net income was $274,998. Interest expenses amounted to $26,878 and $15,468 for the years ended March 31, 2024 and 2023, respectively. The Company recorded a legal settlement expense of $210,500 for the year ended March 31, 2024 and a gain on debt extinguishment of $12,465 and $294,383 for the years ended March 31, 2024 and 2023, respectively.
Net loss
For the years ended March 31, 2024 and 2023, our net loss was $6,739,120 and $5,457,763, respectively. As discussed above, the net loss for the year ended March 31, 2024 and 2023 consists of the expenses discussed above.
46
B. Liquidity and capital resources.
Cash Flows
For the years ended March 31, 2024 and 2023
|
|
For the Year Ended March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Cash used in operating activities |
|
|
(6,247,016 |
) |
|
|
(4,179,767 |
) |
Cash used in investing activities |
|
|
(1,164,449 |
) |
|
|
(178,403 |
) |
Cash provided by financing activities |
|
|
1,648,171 |
|
|
|
13,688,952 |
|
|
|
|
|
|
|
|
||
Change in cash during the year |
|
|
(5,763,294 |
) |
|
|
9,330,782 |
|
Cash, beginning of the year |
|
|
9,352,538 |
|
|
|
21,756 |
|
Cash, end of the year |
|
|
3,589,244 |
|
|
|
9,352,538 |
|
Cash used in operating activities
Net cash used in operating activities was $6,247,016 and $4,179,767 for the years ended March 31, 2024 and 2023, respectively. The increase in cash used for operations was mainly due to increased losses as the Company increased general and administrative and research and development costs associated with developing our Virax brands during the years ended March 31, 2024 and 2023 as discussed above in Results of Operations.
Cash used in investing activities
Net cash used in investing activities was $1,164,449 and $178,403 for the years ended March 31, 2024 and 2023, respectively. Investing activities for the years ended March 31, 2024 and 2023 consisted of $211,952 and $178,403, respectively, of capitalization of certain intangible software costs associated with the development of the ViraxImmune mobile application, which were subsequently impaired. In addition, there were $49,771 of purchases of external software, and $952,497 of purchases of laboratory and computer equipment for the Company's Glasgow laboratory.
Cash provided by financing activities
On July 25, 2022, the Company consummated its IPO of 135,000 ordinary shares, par value $0.001 per share at a price of $50.00 per share. In addition, on July 25, 2022, Boustead Securities, LLC, as representative of several underwriters, exercised an over-allotment option (the “Option”) in part to purchase 20,250 ordinary shares from the Company in connection with the IPO at a price of $50.00 per ordinary share.
The aggregate gross proceeds of our IPO were $7,762,500. After subtracting underwriting discounts and commissions of $543,375 and offering expenses of $169,469, we received net proceeds of approximately $7,049,656.
On November 8, 2022, the Company entered into a Securities Purchase Agreement (the “November SPA”) with an accredited investor (the “Purchaser”) for a private placement offering, pursuant to which the Company received gross proceeds of approximately $3,844,500, before deducting placement agent fees and other offering expenses, in consideration of (i)116,500 Ordinary Shares; (b) 116,500 pre-funded warrants (“November Pre-Funded Warrants”), and (iii) 349,500 warrants (“Ordinary Warrants”) at a combined purchase price of $16.50 per Ordinary Share and one and a half Ordinary Warrant, or approximately $16.50 per Pre-Funded Warrant and one and a half Ordinary Warrant if purchasing the Pre-Funded Warrants (the “November Offering”). The Company has agreed to issue to the Purchaser unregistered warrants to purchase up to 349,500 ordinary shares (the “Ordinary Warrants”).
On March 8, 2023, the Company entered into a second Securities Purchase Agreement (the “Second Securities Purchase Agreement”) with the same Purchaser for a second private placement offering, pursuant to which the Company received gross proceeds of approximately $4,000,000, before deducting placement agent fees and other offering expenses, in consideration of (i)150,000 Ordinary Shares; (b) pre-funded warrants to purchase 234,331 Ordinary Shares (the “March Pre-Funded Warrants”), (iii) series A preferred investment options to purchase up to 349,742 Ordinary Shares (the “Series A Preferred Investment Options”), and (iv) series B preferred investment options to purchase up to 384,331 Ordinary Shares (the “Series B Preferred Investment Options” collectively with the Series A Preferred Investment Options, the “Preferred Options”) at a purchase price of $10.4077 per Ordinary Share and associated Preferred Options and a purchase price of $10.4067 per March Pre-Funded Warrant and associated Preferred Options (the “Offering”). In addition, the Company issued warrants to purchase up to 26,903 Ordinary Shares at $13.010 per share to H.C. Wainwright & Co., placement agent of the Offering, or its assignee (the “Placement Agent Warrants”).
47
On October 11, 2023, the “Company entered into an inducement offer letter agreement (the “Inducement Letter”) with a certain holder (the “Holder”) of 734,073 existing Series A and B preferred investment options (the “Existing Warrants”) to purchase ordinary shares of the Company. The Existing Warrants were issued on March 10, 2023 and each has an exercise price of $8.0202 per share.
Pursuant to the Inducement Letter, the Holder agreed to exercise for cash its Existing Warrants to purchase an aggregate of 734,073 ordinary shares of the Company at a reduced exercise price of $2.934 per share in consideration for the Company’s agreement to issue new warrants to purchase ordinary shares (the “New Warrants”), as described below, to purchase up to 1,468,145 ordinary shares with an exercise price of $2.924 (the “New Warrant Shares”). The Company received aggregate net proceeds of approximately $1.9 million from the exercise of the Existing Warrants by the Holder, after deducting placement agent fees and other offering expenses payable by the Company.
In addition, the Company issued warrants (“Placement Agent Warrants”) to the Placement Agent, or its designees, to purchase up to an aggregate of 51,385 ordinary shares, which Placement Agent Warrants shall be in the form of the New Warrants, except that the Placement Agent Warrants shall have an exercise price of $3.6675 per share.
On January 22, 2024, the Company entered into an At The Market Offering Agreement (the “Sales Agreement”) with H.C. Wainwright & Co., LLC (the “Sales Agent”), acting as the Company’s sales agent, pursuant to which the Company may offer and sell, from time to time, through the Sales Agent, its ordinary shares, having an aggregate offering amount of up to $1,455,029.
Net cash provided by financing activities was $1,648,171 and $13,688,952 for the years ended March 31, 2024 and 2023, respectively. Cash flows from financing activities for the year ended March 31, 2024 were primarily $1.9 million in net proceeds received from the exercise of 734,072 warrants at an exercise price of $2.934. Cash flows used in financing activities were primarily $146,250 in payments on the note payable for the directors and officers insurance offset by stock issued for the settlement of debt of $85,500.
The Company has an accumulated deficit of $18,527,997 at March 31, 2024. We have not generated consistent cash flows to fund our operations yet and as of March 31, 2024, the Company had a cash balance of $3,589,244.
We plan to support our future research and development program, obtain product certification approvals in the territories we have identified, establish our distribution networks, and our general working capital and expenses requirements from our current cash balance. We may, however, over the longer term require additional capital to fund further research and development expenditures and commercialize our products.
At present, we have not generated any significant revenue from existing operations. Our continued existence is dependent on our current cash balance, the ability to obtain necessary financing to fund working capital, complete the planned product certification approvals in the territories we have identified and to establish our distribution networks. We do not expect to generate sufficient internal cash flows to finance these costs in the foreseeable future.
As noted above, the continuation of our current business plan requires us to raise significant additional capital. If we are unable to do so, we may have to curtail our business plans. We intend to use our current cash balance for primarily research and development program, obtaining product certification approvals in the territories we have identified, establishing our distribution networks and for general working capital and expenses purposes.
We will continually evaluate our business plans to determine the manner in which we can most effectively utilize our limited working capital resources. The timing of completion of all aspects of our business plan is highly dependent upon the availability of capital to implement each aspect of the business plan as well as other factors beyond our control.
If our future cash is insufficient to meet our requirements, we may further to seek government grants, to issue debt or equity securities or obtain additional credit facilities. To the extent additional funding is not achieved this will delay our business plans.
C. Research and development, patents and licenses, etc.
For information concerning our research and development policies for the last two years and a description of the amount spent during the last two fiscal years on company-sponsored research and development activities, see “Item 5. Operating and Financial Review and Prospects— Results of Operation.”
D. Trend information.
We are a development stage company and it is not possible for us to predict with any degree of accuracy the outcome of our research, development or commercialization efforts. As such, it is not possible for us to predict with any degree of accuracy any known trends, uncertainties, demands, commitments or events that are reasonably likely to have a material effect on our net sales or revenues, income from continuing operations, profitability, liquidity or capital resources, or that would cause reported financial information to not necessarily be indicative of future operating results or financial conditions. However, to the extent possible, certain trends, uncertainties, demands, commitments and events are in this “Operating and Financial Review and Prospects.”
48
E. Critical Accounting Estimates
We prepare our financial statements in accordance with IFRS. In doing so, we must make estimates and assumptions that affect our reported amounts of assets, liabilities and expenses, as well as related disclosure of contingent assets and liabilities. In some cases, we could reasonably have used different accounting policies and estimates. Changes in the accounting estimates are reasonably likely to occur from period to period. Accordingly, actual results could differ materially from our estimates. To the extent that there are material differences between these estimates and actual results, our financial condition or results of operations will be affected. Significant estimates include, but are not limited to, those related to leases, fixed asset lives, depreciation, intangible asset impairment, and stock-based compensation. For the Company's significant accounting policies, please see Note 2 to our audited consolidated financial statements of this annual report. We believe that our accounting policies contained therein are critical in fully understanding and evaluating our financial condition and operating results.
Item 6. Directors, Senior Management and Employees
A. Directors and senior management.
The following table sets forth information regarding our executive officers and directors as of the date of this Annual Report on form 20-F.
Name |
|
Age |
|
Position |
James Foster |
|
38 |
|
Director, Chief Executive Officer and Chairman |
Nigel McCracken |
|
59 |
|
Chief Operating Officer and Director |
Jason Davis |
|
51 |
|
Chief Financial Officer |
Yair Erez |
|
50 |
|
Independent Director |
Evan Norton |
|
49 |
|
Independent Director |
Nelson Haight |
|
59 |
|
Independent Director |
Below is a summary of the business experience of each our executive officers and directors:
James Foster is our co-founder and has been serving as our Chief Executive Officer, Chairman of the Board of Directors, and Director. From April 2013, Mr. Foster co-founded, served as a board member of Natural Source Group, a product development and distribution company, prior to it merging in creating what became the Virax Biolabs group of Companies. From February 2017 to January 2018, he served as an advisor of Pacific Rim Cobalt Corp. (CSE:BOLT), a publicly traded Electric Vehicle (EV) focused natural resource company. From 2014 to 2015, Mr. Foster served as the co-founder of Cryptex Card Inc., the company that introduces one of the world’s first Bitcoin Debit Card solutions. From 2009 to 2013, he served as co-founder, board member and vice president of Emerging Asia Capital, a resource focused mergers & acquisitions boutique firm. From June 2008 to November 2008, he served within equity sales of NEX Group plc (formerly, ICAP plc), a securities company. From 2004 to 2005, he has worked within fixed income with Royal Bank of Canada. He received a Bachelor’s Degree in American Studies & Chinese from Nottingham University and a Master’s Degree in International Business Management (China) from School of Oriental & African Studies in London in 2008 and 2009, respectively. We believe Mr. Foster’s extensive experience qualifies him to serve as our Chief Executive Officer.
Nigel McCracken is our Chief Operating Officer. Dr. McCracken has been our Chief Operating Officer since September 1, 2023. Prior to joining the Company, from March 2021 to August 2023, Dr. McCracken served as the Chief Scientific Officer of BerGenBio AsA (OTC: BRRGF, LSE: 0RU5 and GR: 7BG), a biopharmaceutical company. From May 2019 to March 2021, Dr. McCracken served as the Chief Operating Officer of NuCana PLC (Nasdaq: NCNA and GR: N04A), a biopharmaceutical company. From September 2014 to April 2019, Dr. McCracken served as the vice president of translational medicine and an executive board member of Debiopharm International SA, a biopharmaceutical company. Dr. McCracken obtained a Master’s degree in Clinical Pharmacology, a Doctor of Philosophy degree in Biochemical Toxicology from Newcastle University, and a Bachelor of Science degree in Biochemistry and Pharmacology from the University of Strathclyde, in 2015, 1991 and 1988, respectively. We believe Dr. McCracken’s extensive experience qualifies him to serve as our Chief Operating Officer.
Jason Davis is our Chief Financial Officer. From December 2019 to December 2021, Mr. Davis served as a vice president of finance of Durango Midstream LLC, a leading natural gas gathering, processing and marketing company providing world-class midstream services to oil and gas producers in Kansas and New Mexico. From February 2017 to November 2019, Mr. Davis served in various consulting roles including interim chief financial officer of Yuma Energy, Inc. (OTC: YUMAQ), a company which explores for and produces crude oil and natural gas, and a vice president of finance and treasurer of Hyperdynamics Corporation (OTC: HDYNQ), an independent oil and gas exploration company. From June 2015 to January 2017, Mr. Davis served as the chief financial officer of Casa Exploration, LLC, an exploration & production company focused on frontier basins in Latin America. Mr. Davis received a Bachelor of Business Administration degree in accounting from the University of Houston in 1997, respectively. Mr. Davis is a certified public accountant in Texas since 1999. We believe Mr. Davis’s extensive experience qualifies him to serve as our Chief Financial Officer.
49
Yair Erez is our independent Director. Since October 2019, Mr. Erez has been a partner at Bain & Co., a consulting firm, focusing on private equity practice and healthcare and life sciences transactions. Since August 2019, Mr. Erez has been the founder of InseytAI Ltd., a Swiss based Artificial Intelligence and Machine Learning company. Since February 2019, Mr. Erez has been a co-founder of Meiji Kickboxing, a chain of kickboxing clubs based in London, United Kingdom. From February 2009 to July 2019, Mr. Erez served as an associate, and subsequently an associate partner, with his final position as a partner of McKinsey & Co., a consulting firm, focusing on private equity, healthcare and life sciences transactions, and growth strategy work for specialty pharma and other life sciences organizations. From 2008 to 2009, Mr. Erez served as the chief executive officer of Tactile World, a company which manufactures assistive technology for blind people. From 2004 to 2008, Mr. Erez served as a senior resident in Obstetrics & Gynecology at Hadassah Ein-Kerem University Hospital, Jerusalem. From 1999 to 2004, he was a major with the Israel Defense Forces. Mr. Erez received a doctor of medicine’s degree from Hebrew University and an executive master of business administration’s degree from Herzelliya Interdisciplinary Center in 1998 and 2010, respectively. We believe Mr. Erez’s extensive experience qualifies him to serve as our independent director.
Evan Norton is our independent Director. Since December 2019, Mr. Norton has been a managing partner at Ballast Capital LLC, a private equity firm. Since September 2016, Mr. Norton has been an adjunct lecturer at Kellogg School of Management of Northwestern University. From November 2019 to May 2021, Mr. Norton served a general partner of Accelmed Partners II L.P., a private equity firm focused on investments in commercial stage Healthtech companies. From January 2010 to November 2019, Mr. Norton served as a director of venture investments and subsequently as managing director of Abbott Laboratories, with his final position as divisional vice president of venture investments of Abbott Laboratories (NYSE: ABT), a medical devices and health care company which provides pharmaceuticals and health care products and services. From 2007 to 2010, Mr. Norton served as a principal of Onset Ventures, a private equity firm which provides early-stage venture capital in the areas of information technology and medical. From 2006 to 2007, Mr. Norton served as a marketing manager of Lifescan, Inc., a subsidiary of Johnson & Johnson (NYSE: JNJ) which focuses on manufacturing products on the diabetes market, specifically blood glucose monitoring systems. From 2002 to 2003, Mr. Norton served a product manager of Stryker Corporation (NYSE: SYK), a medical technologies corporation. From 1998 to 2000, Mr. Norton served as an investment banking associate of JPMorgan Chase & Co. (NYSE: JPM), an investment bank and financial services holding company. From 1996 to 1998, Mr. Norton served as a management consultant in the consulting department of PricewaterhouseCoopers LLP, a public accounting company. Mr. Norton received a master of business administration’s degree from Northwestern University and a bachelor’s degree in business administration in finance from Texas A&M University in 1996 and 2002, respectively. We believe Mr. Norton’s extensive experience qualifies him to serve as our independent director.
Nelson Haight is our independent Director. Mr. Haight is a finance executive with over 30 years of professional experience, Mr. Nelson Haight currently serves as Executive Vice President and Chief Financial Officer of TEAM, Inc., (NYSE: TISI) which he joined in June 2022. Previously from June 2020 to June 2022, he served as Senior Vice President, Chief Financial Officer and Treasurer for Key Energy Services, Inc.. From September 2019 to June 2020, Mr. Haight was the interim Chief Financial Officer for Element Markets, LLC, an environmental commodities firm. From November 2018 to June 2019, Mr. Haight was the interim Chief Financial Officer for Epic Companies, LLC, a family office backed oilfield service company. Between July 2017 and September 2018, Mr. Haight was the Chief Financial Officer of Castleton Resources, LLC, a privately held exploration and production company. From December 2011 to July 2017, Mr. Haight served in various capacities from Vice President to Chief Financial Officer at Midstates Petroleum Company, Inc. Mr. Haight served as a member of the board of directors of Mountain Crest Acquisition Corp (Nasdaq: MCAC) from January 2020 to February 2021, and served as a member of the board of directors of Mountain Crest Acquisition Corp. II (Nasdaq: MCAD) from October 2020 to October 2021. He served as a member of the board of directors of Mountain Crest Acquisition Corp. III (Nasdaq: MCAE) from March 2021 to February 2023. He served as a member of the board of directors of Mountain Crest Acquisition Corp. IV (Nasdaq: MCAF) from March 2021 to March 2024. He has also been serving as a member of the board of directors of Mountain Crest Acquisition Corp. V (Nasdaq: MCAG) since April 2021. Mr. Haight received an MPA and BBA from the University of Texas at Austin in May 1988 and was a licensed Certified Public Accountant up until 2022. We believe Mr. Haight's extensive experience qualifies him to serve as our independent director.
B. Compensation.
Compensation of Directors and Senior Management
The term ‘office holder’ as defined in the Companies Law includes a general manager, chief business manager, deputy general manager, vice general manager, any other person fulfilling or assuming the responsibilities of any of the foregoing positions without regard to such person’s title, as well as a director, or a manager directly subordinate to the general manager or the chief executive officer. As of March 31, 2024, in addition to the three independent members of the Board of Directors, the Company considers two other individuals, to be office holders.
50
The following table presents information regarding compensation reflected in our financial statements for five most highly compensated office holders, as of March 31, 2024.
|
|
Year |
|
Salary ($) |
|
|
Bonus ($) |
|
|
Option Awards ($) (1) |
|
|
Other ($) (2) |
|
|
Total ($) |
|
|||||
James Foster, Chief Executive Officer and Director |
|
2024 |
|
|
325,000 |
|
|
|
81,000 |
|
|
|
241,200 |
|
|
|
3,600 |
|
|
|
650,800 |
|
|
|
2023 |
|
|
247,500 |
|
|
|
213,800 |
|
|
|
1,277,675 |
|
|
|
— |
|
|
|
1,738,975 |
|
|
|
2022 |
|
|
137,766 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
137,766 |
|
Cameron Shaw, Former Chief Operating Officer (3) |
|
2024 |
|
|
150,000 |
|
|
|
— |
|
|
|
— |
|
|
|
93,462 |
|
|
|
243,462 |
|
|
|
2023 |
|
|
215,000 |
|
|
|
205,000 |
|
|
|
1,277,675 |
|
|
|
— |
|
|
|
1,697,675 |
|
|
|
2022 |
|
|
60,000 |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
60,000 |
|
|
Nigel McCracken, Chief Operating Officer and Director (3) |
|
2024 |
|
|
175,000 |
|
|
|
50,000 |
|
|
|
71,600 |
|
|
|
3,540 |
|
|
|
300,140 |
|
|
|
2023 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
2022 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Dr. Tomasz George, Chief Scientific Officer (4) |
|
2024 |
|
|
144,000 |
|
|
|
— |
|
|
|
60,300 |
|
|
|
1,672 |
|
|
|
205,972 |
|
|
|
2023 |
|
|
144,000 |
|
|
|
86,000 |
|
|
|
257,250 |
|
|
|
— |
|
|
|
487,250 |
|
|
|
2022 |
|
|
166,275 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
166,275 |
|
Mark Ternouth, Chief Technical Officer and Director (5) |
|
2024 |
|
|
120,000 |
|
|
|
— |
|
|
|
60,300 |
|
|
|
— |
|
|
|
180,300 |
|
|
|
2023 |
|
|
90,000 |
|
|
|
30,000 |
|
|
|
315,560 |
|
|
|
— |
|
|
|
435,560 |
|
|
|
2022 |
|
|
11,200 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
11,200 |
|
Jason Davis, Chief Financial Officer |
|
2024 |
|
|
300,000 |
|
|
|
75,000 |
|
|
|
224,318 |
|
|
|
25,800 |
|
|
|
625,118 |
|
|
|
2023 |
|
|
237,500 |
|
|
|
105,000 |
|
|
|
686,000 |
|
|
|
— |
|
|
|
1,028,500 |
|
|
|
2022 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Evan Norton, Independent Director |
|
2024 |
|
|
40,000 |
|
|
|
— |
|
|
|
60,300 |
|
|
|
— |
|
|
|
100,300 |
|
|
|
2023 |
|
|
27,500 |
|
|
|
— |
|
|
|
68,600 |
|
|
|
— |
|
|
|
96,100 |
|
|
|
2022 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Yair Erez, Independent Director |
|
2024 |
|
|
40,000 |
|
|
|
— |
|
|
|
60,300 |
|
|
|
— |
|
|
|
100,300 |
|
|
|
2023 |
|
|
27,500 |
|
|
|
— |
|
|
|
68,600 |
|
|
|
— |
|
|
|
96,100 |
|
|
|
2022 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Nelson Haight, Independent Director |
|
2024 |
|
|
40,000 |
|
|
|
— |
|
|
|
60,300 |
|
|
|
— |
|
|
|
100,300 |
|
|
|
2023 |
|
|
14,783 |
|
|
|
— |
|
|
|
27,400 |
|
|
|
— |
|
|
|
42,183 |
|
|
|
2022 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
(1) These amounts represent the aggregate grant fair value of stock options granted in the year ended March 31, 2024 and 2023, calculated in accordance with IFRS 2 “Share-based payment". Assumptions used in the calculation of these amounts are discussed in Note 18 to our Consolidated Audited Financial Statements for the year ended March 31, 2024 and March 31, 2023, included in Item 8. Financial Information, below.
(2)These amounts represent employee benefits paid on behalf of the Company such as pension and health insurance. For Cameron Shaw, this amount consists of vacation and severance payout.
(3) Cameron Shaw departed the Company and Nigel McCracken joined the Company on September 1, 2023.
(4) Dr. Tomasz George departed the Company on April 16, 2024.
(5) Mark Ternouth departed the Company on June 11, 2024.
Employment Agreements
We have entered into employment agreements with each of our executive officers. We may terminate the employment for cause at any time for certain acts, such as conviction or plea of guilty to a felony or any crime involving moral turpitude, negligent or dishonest acts to our detriment, or misconduct or a failure to perform agreed duties. Termination of employment without cause varies between 3 months’ advance written notice to 12 months' advance written notice.. Each executive officer may resign at any time.
Each executive officer has agreed to hold, both during and after the termination or expiry of his employment agreement, in strict confidence and not to use, except as required in the performance of his duties in connection with the employment or pursuant to applicable law, any of our confidential or proprietary information or the confidential or proprietary information of any third party received by us and for which we have confidential obligations. Each executive officer has also agreed to disclose in confidence to us all inventions, designs and trade secrets which he conceives, develops or reduces to practice during his employment with us and to
51
assign all right, title and interest in them to us, and assist us in obtaining and enforcing patents, copyrights and other legal rights for these inventions, designs and trade secrets.
In addition, each executive officer has agreed to be bound by non-competition and non-solicitation restrictions during the term of the employment and for one year following the last date of employment. Specifically, each executive officer has agreed not to: (i) engage or assist others in engaging in any business or enterprise that is competitive with our business, (ii) solicit, divert or take away the business of our clients, customers or business partners, or (iii) solicit, induce or attempt to induce any employee or independent contractor to terminate his or her employment or engagement with us. The employment agreements also contain other customary terms and provisions.
We have also entered into director agreements with each of our directors which agreements set forth the terms and provisions of their engagement.
Each of our executive's employment agreements are filed as an exhibit in this Annual Report on Form 20-F.
Equity Incentive Plans
Our Board and shareholders adopted a 2022 Equity Incentive Plan and a 2023 Equity Incentive Plan to provide additional means through the grant of awards to attract, motivate, retain and reward selected key employees and other eligible persons. Below is a summary of the equity incentive plans terms:
Shares Subject to the equity incentive plans
A total of 39,491 of our ordinary shares is available for issuance under the 2022 Equity Incentive Plan and a total of 100,250 of our ordinary shares is available for issuance under the 2023 Equity Incentive Plan. If an award granted under either equity incentive plan is forfeited, canceled, settled, or otherwise terminated without a distribution of ordinary shares, the ordinary shares underlying that award will again become available for issuance under the equity incentive plan. If ordinary shares delivered under the 2022 Equity Incentive Plan or 2023 Equity Incentive Plan are tendered or withheld to pay the exercise price of a share option or to satisfy withholding taxes, those ordinary shares will also again become available for issuance under either of the equity incentive plans.
Administration of the equity incentive plans
Our Board or a committee appointed by the Board will administer the equity incentive plans. The plan administrator will have broad authority to:
Participation
Employees, directors and consultants that provide services to us or one of our subsidiaries may be selected to receive awards under the equity incentive plans.
Types of Awards
The equity incentive plans permit the granting of awards in the form of share options, performance awards, or other awards.
Share Options
A share option entitles the recipient to purchase ordinary shares at a fixed exercise price. The exercise price per share will be determined by the plan administrator in the applicable award agreement in its sole discretion at the time of the grant, but the exercise price cannot
52
be less than the closing sales price for our ordinary shares on the grant date. The exercise price can be paid in cash, check, or by cashless or net exercise. The maximum term of each share option shall be fixed by the plan administrator, but in no event shall an option be exercisable more than ten (10) years after the date such option is granted.
Performance Awards
A performance award is an award that may vest contingent upon the attainment during a performance period of certain performance goals and which is granted under the terms and conditions of other awards pursuant to such terms and conditions established by the plan administrator.
Equitable Adjustments
In the event of a merger, consolidation, recapitalization, share split, reverse share split, reorganization, split-up, spin-off, combination, repurchase, or other change in corporate structure affecting the ordinary shares, the maximum number and kind of shares reserved for issuance or with respect to which awards may be granted under the equity incentive plan will be adjusted to reflect such event, and the plan administrator will make such adjustments as it deems appropriate and equitable in the number, kind and exercise price of ordinary shares covered by outstanding awards made under the equity incentive plan.
Change in Control
In the event of any proposed change in control (as defined in the equity incentive plans), the plan administrator will take any action as it deems appropriate, which action may include, without limitation, the following: (i) the continuation of any award, if the company is the surviving corporation; (ii) the assumption of any award by the surviving corporation or its parent or subsidiary; (iii) the substitution by the surviving corporation or its parent or subsidiary of equivalent awards; (iv) accelerated vesting of the award, with all performance objectives and other vesting criteria deemed achieved at targeted levels, and a limited period during which to exercise the award prior to closing of the change in control, or (v) cash settlement equal to the fair market value of the shares that would otherwise be issued to the recipient.
Term
The equity incentive plans will become effective when adopted by the Board and, unless terminated, the equity incentive plans will continue in effect for a term of ten (10) years.
Amendment and Termination
The Board may at any time amend, alter, suspend or terminate the equity incentive plan, although no such action may, without the written consent of the participant, impair the rights of any participant with respect to outstanding awards.
Status
Total equity awards outstanding under the 2022 Equity Incentive Plan are 131,941 and there are 250,000 awards outstanding under the 2023 Equity Incentive Plan as of March 31, 2024.
C. Board Practices
Composition of our Board of Directors
Our board of directors consists of five directors. A director is not required to hold any shares in our company to qualify to serve as a director. The Corporate Governance Rules of the Nasdaq generally require that a majority of an issuer’s board of directors must consist of independent directors.
Our board of directors currently consists of five directors. Our board of directors has determined that each of Mr. Erez, Mr. Norton, and Mr. Haight is an “independent director” as defined under the Nasdaq rules. Our board of directors is composed of a majority of independent directors.
A director is not required to hold any of our shares to qualify to serve as a director.
Committees of the Board of Directors
We have established an audit committee, a compensation committee and a nominating and corporate governance committee under our Board of Directors. We have adopted a charter for each of the three committees. Each committee’s members and functions are described below.
53
Audit Committee.
Our audit committee consists of our three independent directors and is chaired by Mr. Haight. We have determined that satisfy the requirements of Section 303A of the Corporate Governance Rules/ Rule 5605(c)(2) of the Listing Rules of the NASDAQ and meet the independence standards under Rule 10A-3 under the Securities Exchange Act of 1934, as amended. We have determined that qualifies as an “audit committee financial expert.” The audit committee oversees our accounting and financial reporting processes and the audits of the financial statements of our company. The audit committee is responsible for, among other things:
Compensation Committee.
Our compensation committee consists of our three independent directors and is chaired by Mr. Erez. We have determined that satisfy the “independence” requirements of Rule5605(c)(2) of the Listing Rules of the NASDAQ. The compensation committee assists the board in reviewing and approving the compensation structure, including all forms of compensation, relating to our directors and executive officers. Our chief executive officer may not be present at any committee meeting during which their compensation is deliberated upon. The compensation committee is responsible for, among other things:
Nominating and Corporate Governance Committee.
Our nominating and corporate governance committee consists of our three independent directors, and is chaired by Mr. Norton. We have determined that satisfy the “independence” requirements of Rule5605(c)(2) of the Listing Rules of the NASDAQ. The nominating and corporate governance committee assists the board in selecting individuals qualified to become our directors and in determining the composition of the board and its committees. The nominating and corporate governance committee is responsible for, among other things:
54
Code of Business Conduct and Ethics
We have adopted a code of business conduct and ethics, which is applicable to all of our directors, executive officers and employees and is publicly available.
Duties of Directors
Under Cayman Islands law, our board of directors has the powers necessary for managing, and for directing and supervising, our business affairs. The functions and powers of our board of directors include, among others:
Under Cayman Islands law, directors owe the following fiduciary duties: (i) duty to act in good faith in what the director believes to be in the best interests of the company as a whole; (ii) duty to exercise powers for the purposes for which those powers were conferred and not for a collateral purpose; (iii) directors should not improperly fetter the exercise of future discretion; (iv) duty not to put themselves in a position in which there is a conflict between their duty to the company and their personal interests; and (v) duty to exercise independent judgment. In addition to the above, directors also owe a duty to act with skill, care and diligence. This duty has been defined as a requirement to act as a reasonably diligent person having both the general knowledge, skill and experience that may reasonably be expected of a person carrying out the same functions as are carried out by that director in relation to the company and the general knowledge skill and experience which that director has.
As set out above, directors have a duty not to put themselves in a position of conflict and this includes a duty not to engage in self-dealing, or to otherwise benefit as a result of their position. However, in some instances what would otherwise be a breach of this duty can be forgiven and/or authorized in advance by the shareholders provided that there is full disclosure by the directors. This can be done by way of permission granted in the memorandum and articles of association or alternatively by shareholder approval at general meetings.
D. Employees
As of March 31, 2024, we had 17 full-time employees. There were 11 employees in the United Kingdom, 1 employee in the United States, and 5 employees in China.
E. Share Ownership
See “Item 7.A. Major Shareholders and Related Party Transactions – Major Shareholders.” Our employees are eligible to own shares of the company through a warrant incentive plan. For information on the plan, see “Item 6.B. Compensation—Equity Incentive Plan.”
Item 7. Major Shareholders and Related Party Transactions
A. Major Shareholders
55
The following table presents information, as of the date of this report, regarding the beneficial ownership of our ordinary shares by:
The number of ordinary shares beneficially owned by each entity, person, and member of our board of directors or members of our executive management is determined in accordance with the rules of the SEC, and the information is not necessarily indicative of beneficial ownership for any other purpose. Under such rules, beneficial ownership includes any ordinary shares over which the individual has sole or shared voting power or investment power as well as any ordinary shares that the individual has the right to acquire within 60 days of the date of this report through the exercise of any option, warrant or other right. Except as otherwise indicated, and subject to applicable community property laws, the persons named in the table have sole voting and investment power with respect to all ordinary shares held by that person.
The percentage of outstanding ordinary shares is computed on the basis of 2,964,920 ordinary shares outstanding as of the date of this report. Ordinary shares that a person has the right to acquire within 60 days of the date of this report are deemed outstanding for purposes of computing the percentage ownership of the person holding such rights but are not deemed outstanding for purposes of computing the percentage ownership of any other person, except with respect to the percentage ownership of all members of our board of directors or executive management as a group. None of our shareholders has different voting rights from other shareholders. We are not aware of any arrangement that may, at a subsequent date, result in a change of control of our company.
Names of beneficial owners(1) |
|
Number of ordinary shares |
|
Percentage of class (%) |
|
|||||
|
|
|
|
|
|
|
|
|
||
James Foster |
|
|
287,987 |
|
|
(2) |
|
|
11.2 |
% |
|
|
|
|
|
|
|
|
|
||
Jason Davis |
|
|
— |
|
|
|
|
* |
|
|
|
|
|
|
|
|
|
|
|
||
Nigel McCracken |
|
|
— |
|
|
|
|
* |
|
|
|
|
|
|
|
|
|
|
|
||
Nelson Haight |
|
|
— |
|
|
|
|
* |
|
|
|
|
|
|
|
|
|
|
|
||
Yair Erez |
|
|
— |
|
|
|
|
* |
|
|
|
|
|
|
|
|
|
|
|
||
Evan Norton |
|
|
— |
|
|
|
|
* |
|
|
|
|
|
|
|
|
|
|
|
||
All officers and directors as a group (six (6) persons) |
|
|
287,987 |
|
|
|
|
|
11.2 |
% |
|
|
|
|
|
|
|
|
|
||
There are no other 5% or greater shareholders |
|
|
|
|
|
|
|
|
||
* Denotes less than 1%
(1) Unless otherwise noted, the business address of each of the following entities or individuals is Bo’Ness Road Newhouse, Lanarkshire, ML1 5UH.
(2) Consists of ordinary shares owned by the individual.
B. Related Party Transactions
There were no loans between the Company and related parties for the period since the beginning of the Company’s preceding three financial years up to the date of the filing of the annual report on Form 20-F. Transactions such as stock-based compensation, reimbursements and payroll related items are discussed in Item 6.B in this report.
C. Interests of experts and counsel
Not applicable.
Item 8. Financial Information
A. Consolidated Statements and Other Financial Information.
See “Item 18. Financial Statements” for a list of all financial statements filed as part of this Annual Report on Form 20-F.
Legal Matters
We are not involved in any legal or arbitration proceedings that may have or have had in the recent past, significant effects on our financial position or profitability.
56
Dividend Policy
We have never declared or paid cash dividends to our shareholders. Currently we do not intend to pay cash dividends. We intend to reinvest any earnings in developing and expanding our business. Any future determination relating to our dividend policy will be at the discretion of our Board of Directors and will depend on a number of factors, including future earnings, our financial condition, operating results, contractual restrictions, capital requirements, business prospects, applicable Cayman Companies Law and other factors our Board of Directors may deem relevant.
B. Significant Changes.
See “Note 19 - Subsequent Events” to our consolidated financial statements included in this Annual Report on Form 20-F beginning on page F-1 for a discussion of significant events that have occurred since March 31, 2024.
Item 9. The Offer and Listing.
A. Offer and listing details.
ordinary shares
Our ordinary shares have been trading on the Nasdaq under the symbol “VRAX” since July 21, 2022.
B. Plan of distribution.
Not applicable.
C. Markets.
See “—Offer and Listing Details” above.
D. Selling shareholders
Not applicable.
E. Dilution.
Not applicable
F. Expenses of the issue.
Not applicable.
Item 10. Additional Information
A. Share capital.
Not applicable.
B. Memorandum and articles of association.
We are an exempted company incorporated with limited liability under the laws of the Cayman Islands and our affairs are governed by:
Our authorized share capital is US$50,000 divided into 50,000,000 ordinary shares of $0.001 par value each.
57
We have included summaries of certain material provisions of our second amended and restated memorandum and articles of association (the Memorandum and Articles, respectively) and the Cayman Companies Act insofar as they relate to the material terms of our share capital. The summaries do not purport to be complete and are qualified in their entirety by reference to our Memorandum and Articles, which is filed as Exhibit 1.1 to this annual report.
Ordinary shares
All of our outstanding ordinary shares are fully paid and non-assessable. Certificates representing the ordinary shares are issued in registered form. Our shareholders, whether or not they are non-residents of the Cayman Islands, may freely hold and transfer their ordinary shares in accordance with our Memorandum and Articles.
Dividends
The holders of our ordinary shares are entitled to such dividends as may be declared by our board of directors. Our Articles provide that our board of directors may declare and pay dividends if justified by our financial position and permitted by law. Our articles of association also provides that, subject to the Cayman Companies Act, the Company may also by ordinary resolution declare dividends in accordance with the respective rights of the shareholders but no dividend shall exceed the amount recommended by the directors.
Voting Rights
Holders of our ordinary shares vote on all matters submitted to a vote of our shareholders, except as may otherwise be required by law. In respect of matters requiring shareholders’ vote, each ordinary share is entitled to one vote. At any general meeting a resolution put to the vote of the meeting shall be decided on a show of hands unless voting by poll is duly demanded by the chairman of the meeting, by at least two shareholders having the right to vote on the resolutions, or by shareholder(s) together holding at least 10% of the total voting rights of all our shareholders having the right to vote at such general meeting. A quorum required for a meeting of shareholders consists of one or more shareholders who holds at least one-third of our issued voting shares. Shareholders’ meetings may be held annually. Each general meeting, other than an annual general meeting, shall be an extraordinary general meeting. Extraordinary general meetings may be called by a majority of our board of directors or upon a requisition of any one or more shareholders holding at the deposit of the requisition at least 10% of the aggregate share capital of our company that carries the right to vote at a general meeting, in which case on advance notice of at least 7 clear days is required for the convening of our annual general meeting and other general meetings by requisition of our shareholders.
Any ordinary resolution to be made by the shareholders requires the affirmative vote of a simple majority of the votes attaching to the ordinary shares cast in a meeting, while a special resolution requires the affirmative vote of no less than two-thirds of the votes attaching to the ordinary shares cast in a meeting.
A special resolution will be required for important matters such as amending our memorandum and articles of association or changing the name of the Company.
There are no limitations on non-residents or foreign shareholders in the memorandum and articles of association to hold or exercise voting rights on the ordinary shares imposed by foreign law or by the charter or other constituent document of our company. However, no person will be entitled to vote at any general meeting or at any separate meeting of the holders of the ordinary shares unless the person is registered as of the record date for such meeting and unless all calls or other sums presently payable by the person in respect of ordinary shares in the Company have been paid.
Winding Up; Liquidation
Subject to any special rights, privileges or restrictions as to the distribution of available surplus assets on liquidation applicable to any class or classes of shares (1) if we are wound up and the assets available for distribution among our shareholders are more than sufficient to repay the whole of the capital paid up at the commencement of the winding up, the excess shall be distributed pari passu among our shareholders in proportion to the amount paid up at the commencement of the winding up on the shares held by them, respectively, and (2) if we are wound up and the assets available for distribution among our shareholders as such are insufficient to repay the whole of the paid-up capital, those assets shall be distributed so that, as nearly as may be, the losses shall be borne by our shareholders in proportion to the capital paid up, or which ought to have been paid up, at the commencement of the winding up on the shares held by them, respectively.
Calls on ordinary shares and Forfeiture of ordinary shares
58
Our directors may from time to time make calls on our shareholders in respect of any moneys unpaid on their shares including any premium in a notice served to such shareholders at least 14 clear days prior to the specified time of payment. Any ordinary shares that have been called upon and remain unpaid are subject to forfeiture.
Redemption of ordinary shares
The Cayman Companies Act and our Memorandum and Articles permit us to purchase our own shares. In accordance with our Articles, provided the necessary shareholders or board approval have been obtained and requirements under the Cayman Companies Act have been satisfied, we may issue shares on terms that are subject to redemption at our option on such terms and in such manner as may be determined by our board of directors.
Inspection of Books and Records
Holders of our ordinary shares have no general right under our Articles to inspect or obtain copies of our list of shareholders or our corporate records. However, we will provide our shareholders with annual audited financial statements.
Issuance of Additional Shares
Our Memorandum and Articles authorize our board of directors to issue additional ordinary shares from time to time as our board of directors shall determine, to the extent of available authorized but unissued shares. Issuance of these shares may dilute the voting power of holders of ordinary shares.
Anti-Takeover Provisions
Some provisions of our Memorandum and Articles may discourage, delay or prevent a change of control of our company or management that shareholders may consider favorable. Our authorized, but unissued ordinary shares are available for future issuance without shareholders’ approval and could be utilized for a variety of corporate purposes, including future offerings to raise addition capital, acquisitions and employee benefit plans. The existence of authorized but unissued and unreserved ordinary shares could render more difficult or discourage an attempt to obtain control of us by means of a proxy contest, tender offer, merger or otherwise.
Exempted Company
We are an exempted company with limited liability under the Cayman Companies Act. The Cayman Companies Act distinguishes between ordinary resident companies and exempted companies. Any company that is registered in the Cayman Islands but conducts business mainly outside of the Cayman Islands may apply to be registered as an exempted company. The requirements for an exempted company are essentially the same as for an ordinary company except that an exempted company:
“Limited liability” means that the liability of each shareholder is limited to the amount unpaid by the shareholder on the shares of the company.
C. Material contracts.
We have not entered into any material contracts other than in the ordinary course of business and other than those described in “Item 4. Information on the Company” or elsewhere in this annual report on Form 20-F.
59
D. Exchange controls.
There are no government laws, decrees or regulations that restrict or that affect our export or import of capital or the remittance of dividends, interest or other payments to non-resident holders of our securities, including the availability of cash and cash equivalents for use by us and our wholly-owned subsidiary, except or otherwise as set forth under “Item 10. Additional Information—E. Taxation” and “Item 3. Key Information Risk Factors – D. Risk Factors – Risks Related to Doing Business in China and Hong Kong - Restrictions on currency exchange may limit our ability to utilize our revenues effectively”.
E. Taxation.
The following discussion describes the material U.S. federal income tax consequences relating to the ownership and disposition of our ordinary shares by U.S. Holders (as defined below). This discussion applies to U.S. Holders that purchase our ordinary shares pursuant to the Company's IPO and hold such ordinary shares as capital assets. This discussion is based on the U.S. Internal Revenue Code of 1986, as amended, U.S. Treasury regulations promulgated thereunder and administrative and judicial interpretations thereof, all as in effect on the date hereof and all of which are subject to change, possibly with retroactive effect. This discussion does not address all of the U.S. federal income tax consequences that may be relevant to specific U.S. Holders in light of their particular circumstances or to U.S. Holders subject to special treatment under U.S. federal income tax law (such as certain financial institutions, insurance companies, dealers or traders in securities or other persons that generally mark their securities to market for U.S. federal income tax purposes, tax-exempt entities or governmental organizations, retirement plans, regulated investment companies, real estate investment trusts, grantor trusts, brokers, dealers or traders in securities, commodities, currencies or notional principal contracts, certain former citizens or long-term residents of the United States, persons who hold our ordinary shares as part of a “straddle,” “hedge,” “conversion transaction,” “synthetic security” or integrated investment, persons that have a “functional currency” other than the U.S. dollar, persons that own directly, indirectly or through attribution 10% or more of the voting power of our ordinary shares, corporations that accumulate earnings to avoid U.S. federal income tax, partnerships and other pass-through entities, and investors in such pass-through entities). This discussion does not address any U.S. state or local or non-U.S. tax consequences or any U.S. federal estate, gift or alternative minimum tax consequences.
As used in this discussion, the term “U.S. Holder” means a beneficial owner of our ordinary shares who is, for U.S. federal income tax purposes, (i) an individual who is a citizen or resident of the United States, (ii) a corporation (or entity treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States, any state thereof, or the District of Columbia, (iii) an estate the income of which is subject to U.S. federal income tax regardless of its source or (iv) a trust (x) with respect to which a court within the United States is able to exercise primary supervision over its administration and one or more United States persons have the authority to control all of its substantial decisions or (y) that has elected under applicable U.S. Treasury regulations to be treated as a domestic trust for U.S. federal income tax purposes.
If an entity treated as a partnership for U.S. federal income tax purposes holds our ordinary shares, the U.S. federal income tax consequences relating to an investment in such ordinary shares will depend in part upon the status and activities of such entity and the particular partner. Any such entity should consult its own tax advisor regarding the U.S. federal income tax consequences applicable to it and its partners of the purchase, ownership and disposition of our ordinary shares.
Persons considering an investment in our ordinary shares should consult their own tax advisors as to the particular tax consequences applicable to them relating to the purchase, ownership and disposition of our ordinary shares including the applicability of U.S. federal, state and local tax laws and non-U.S. tax laws.
Passive Foreign Investment Company Consequences
In general, a corporation organized outside the United States will be treated as a PFIC for any taxable year in which either (i) at least 75% of its gross income is “passive income”, or the PFIC income test, or (ii) on average at least 50% of its assets, determined on a quarterly basis, are assets that produce passive income or are held for the production of passive income, or the PFIC asset test. Passive income for this purpose generally includes, among other things, dividends, interest, royalties, rents, and gains from the sale or exchange of property that gives rise to passive income. Assets that produce or are held for the production of passive income generally include cash, even if held as working capital or raised in a public offering, marketable securities, and other assets that may produce passive income. Generally, in determining whether a non-U.S. corporation is a PFIC, a proportionate share of the income and assets of each corporation in which it owns, directly or indirectly, at least a 25% interest (by value) is taken into account.
Although PFIC status is determined on an annual basis and generally cannot be determined until the end of a taxable year, based on the nature of our current and expected income and the current and expected value and composition of our assets, we do not presently expect to be a PFIC for our current taxable year or the foreseeable future. However, there can be no assurance given in this regard because the determination of whether we are or will become a PFIC is a fact-intensive inquiry made on an annual basis that depends, in part, upon
60
the composition of our income and assets. In addition, there can be no assurance that the IRS will agree with our conclusion or that the IRS would not successfully challenge our position.
If we are a PFIC in any taxable year during which a U.S. Holder owns our ordinary shares, the U.S. Holder could be liable for additional taxes and interest charges under the “PFIC excess distribution regime” upon (i) a distribution paid during a taxable year that is greater than 125% of the average annual distributions paid in the three preceding taxable years, or, if shorter, the U.S. Holder’s holding period for our ordinary shares, and (ii) any gain recognized on a sale, exchange or other disposition, including a pledge, of our ordinary shares, whether or not we continue to be a PFIC. Under the PFIC excess distribution regime, the tax on such distribution or gain would be determined by allocating the distribution or gain ratably over the U.S. Holder’s holding period for our ordinary shares. The amount allocated to the current taxable year (i.e., the year in which the distribution occurs or the gain is recognized) and any year prior to the first taxable year in which we are a PFIC will be taxed as ordinary income earned in the current taxable year. The amount allocated to other taxable years will be taxed at the highest marginal rates in effect for individuals or corporations, as applicable, to ordinary income for each such taxable year, and an interest charge, generally applicable to underpayments of tax, will be added to the tax.
If we are a PFIC for any year during which a U.S. Holder holds our ordinary shares, we must generally continue to be treated as a PFIC by that holder for all succeeding years during which the U.S. Holder holds such ordinary shares, unless we cease to meet the requirements for PFIC status and the U.S. Holder makes a “deemed sale” election with respect to our ordinary shares. If the election is made, the U.S. Holder will be deemed to sell our ordinary shares it holds at their fair market value on the last day of the last taxable year in which we qualified as a PFIC, and any gain recognized from such deemed sale would be taxed under the PFIC excess distribution regime. After the deemed sale election, the U.S. Holder’s ordinary shares would not be treated as shares of a PFIC unless we subsequently become a PFIC.
If we are a PFIC for any taxable year during which a U.S. Holder holds our ordinary shares and one of our non-United States subsidiaries is also a PFIC (i.e., a lower-tier PFIC), such U.S. Holder would be treated as owning a proportionate amount (by value) of the shares of the lower-tier PFIC and would be taxed under the PFIC excess distribution regime on distributions by the lower-tier PFIC and on gain from the disposition of shares of the lower-tier PFIC even though such U.S. Holder would not receive the proceeds of those distributions or dispositions. Any of our non-United States subsidiaries that have elected to be disregarded as entities separate from us or as partnerships for U.S. federal income tax purposes would not be corporations under U.S. federal income tax law and accordingly, cannot be classified as lower-tier PFICs. However, non-United States subsidiaries that have not made the election may be classified as a lower-tier PFIC if we are a PFIC during your holding period and the subsidiary meets the PFIC income test or PFIC asset test. Each U.S. Holder is advised to consult its tax advisors regarding the application of the PFIC rules to any of our non-United States subsidiaries.
If we are a PFIC, a U.S. Holder will not be subject to tax under the PFIC excess distribution regime on distributions or gain recognized on our ordinary shares if a valid “mark-to-market” election is made by the U.S. Holder for our ordinary shares. An electing U.S. Holder generally would take into account as ordinary income each year, the excess of the fair market value of our ordinary shares held at the end of such taxable year over the adjusted tax basis of such ordinary shares. The U.S. Holder would also take into account, as an ordinary loss each year, the excess of the adjusted tax basis of such ordinary shares over their fair market value at the end of the taxable year, but only to the extent of the excess of amounts previously included in income over ordinary losses deducted as a result of the mark-to-market election. The U.S. Holder’s tax basis in our ordinary shares would be adjusted to reflect any income or loss recognized as a result of the mark-to-market election. Any gain from a sale, exchange or other disposition of our ordinary shares in any taxable year in which we are a PFIC would be treated as ordinary income and any loss from such sale, exchange or other disposition would be treated first as ordinary loss (to the extent of any net mark-to-market gains previously included in income) and thereafter as capital loss. If, after having been a PFIC for a taxable year, we cease to be classified as a PFIC because we no longer meet the PFIC income or PFIC asset test, the U.S. Holder would not be required to take into account any latent gain or loss in the manner described above and any gain or loss recognized on the sale or exchange of the ordinary shares would be classified as a capital gain or loss.
A mark-to-market election is available to a U.S. Holder only for “marketable stock.” Generally, stock will be considered marketable stock if it is “regularly traded” on a “qualified exchange” within the meaning of applicable U.S. Treasury regulations. A class of stock is regularly traded during any calendar year during which such class of stock is traded, other than in de minimis quantities, on at least fifteen (15) days during each calendar quarter.
Our ordinary shares will be marketable stock as long as they remain listed on the Nasdaq Capital Market and are regularly traded. A mark-to-market election will not apply to the ordinary shares for any taxable year during which we are not a PFIC, but will remain in effect with respect to any subsequent taxable year in which we become a PFIC. Such election will not apply to any of our non-U.S. subsidiaries. Accordingly, a U.S. Holder may continue to be subject to tax under the PFIC excess distribution regime with respect to any lower-tier PFICs notwithstanding the U.S. Holder’s mark-to-market election for the ordinary shares.
The Cayman Islands currently have no form of income, corporate or capital gains tax and no estate duty, inheritance tax or gift tax. There are currently no Cayman Islands’ taxes or duties of any nature on gains realized on a sale, exchange, conversion, transfer or redemption of the ordinary shares. Payments of dividends and capital in respect of the ordinary shares will not be subject to taxation in the Cayman Islands and no withholding will be required on the payment of interest and principal or a dividend or capital to any holder
61
of the ordinary shares, nor will gains derived from the disposal of the ordinary shares be subject to Cayman Islands income or corporation tax as the Cayman Islands currently have no form of income or corporation taxes.
The tax consequences that would apply if we are a PFIC would also be different from those described above if a U.S. Holder were able to make a valid qualified electing fund, or QEF, election. As we do not expect to provide U.S. Holders with the information necessary for a U.S. Holder to make a QEF election, prospective investors should assume that a QEF election will not be available.
The U.S. federal income tax rules relating to PFICs are very complex. Prospective U.S. investors are strongly urged to consult their own tax advisors with respect to the impact of PFIC status on the purchase, ownership and disposition of our ordinary shares, the consequences to them of an investment in a PFIC, any elections available with respect to the ordinary shares and the IRS information reporting obligations with respect to the purchase, ownership and disposition of ordinary shares of a PFIC.
Distributions
Subject to the discussion above under “— Passive Foreign Investment Company Consequences,” a U.S. Holder that receives a distribution with respect to our ordinary shares generally will be required to include the gross amount of such distribution in gross income as a dividend when actually or constructively received to the extent of the U.S. Holder’s pro rata share of our current and/or accumulated earnings and profits (as determined under U.S. federal income tax principles). To the extent a distribution received by a U.S. Holder is not a dividend because it exceeds the U.S. Holder’s pro rata share of our current and accumulated earnings and profits, it will be treated first as a tax-free return of capital and reduce (but not below zero) the adjusted tax basis of the U.S. Holder’s ordinary shares. To the extent the distribution exceeds the adjusted tax basis of the U.S. Holder’s ordinary shares, the remainder will be taxed as capital gain. Because we may not account for our earnings and profits in accordance with U.S. federal income tax principles, U.S. Holders should expect all distributions to be reported to them as dividends.
Distributions on our ordinary shares that are treated as dividends generally will constitute income from sources outside the United States for foreign tax credit purposes and generally will constitute passive category income. Such dividends will not be eligible for the “dividends received’’ deduction generally allowed to corporate shareholders with respect to dividends received from U.S. corporations. Dividends paid by a “qualified foreign corporation’’ to certain non-corporate U.S. Holders may be are eligible for taxation at a reduced capital gains rate rather than the marginal tax rates generally applicable to ordinary income provided that a holding period requirement (more than sixty (60) days of ownership, without protection from the risk of loss, during the 121-day period beginning sixty (60) days before the ex-dividend date) and certain other requirements are met. Each U.S. Holder is advised to consult its tax advisors regarding the availability of the reduced tax rate on dividends to its particular circumstances. However, if we are a PFIC for the taxable year in which the dividend is paid or the preceding taxable year (see discussion above under “— Passive Foreign Investment Company Consequences’’), we will not be treated as a qualified foreign corporation, and therefore the reduced capital gains tax rate described above will not apply.
Dividends will be included in a U.S. Holder’s income on the date of the depositary’s receipt of the dividend. The amount of any dividend income paid in Cayman Islands dollars will be the U.S. dollar amount calculated by reference to the exchange rate in effect on the date of receipt, regardless of whether the payment is in fact converted into U.S. dollars. If the dividend is converted into U.S. dollars on the date of receipt, a U.S. Holder should not be required to recognize foreign currency gain or loss in respect to the dividend income. A U.S. Holder may have foreign currency gain or loss if the dividend is converted into U.S. dollars after the date of receipt.
A non-United States corporation (other than a corporation that is classified as a PFIC for the taxable year in which the dividend is paid or the preceding taxable year) generally will be considered to be a qualified foreign corporation with respect to any dividend it pays on ordinary shares that are readily tradable on an established securities market in the United States.
Sale, Exchange or Other Disposition of Our ordinary shares
Subject to the discussion above under “— Passive Foreign Investment Company Consequences,’’ a U.S. Holder generally will recognize capital gain or loss for U.S. federal income tax purposes upon the sale, exchange or other disposition of our ordinary shares in an amount equal to the difference, if any, between the amount realized (i.e., the amount of cash plus the fair market value of any property received) on the sale, exchange or other disposition and such U.S. Holder’s adjusted tax basis in the ordinary shares. Such capital gain or loss generally will be long-term capital gain taxable at a reduced rate for non-corporate U.S. Holders or long-term capital loss if, on the date of sale, exchange or other disposition, the ordinary shares were held by the U.S. Holder for more than one year. Any capital gain of a non-corporate U.S. Holder that is not long-term capital gain is taxed at ordinary income rates. The deductibility of capital losses is subject to limitations. Any gain or loss recognized from the sale or other disposition of our ordinary shares will generally be gain or loss from sources within the United States for U.S. foreign tax credit purposes.
Medicare Tax
Certain U.S. Holders that are individuals, estates or trusts and whose income exceeds certain thresholds generally are subject to a 3.8% tax on all or a portion of their net investment income, which may include their gross dividend income and net gains from the disposition
62
of our ordinary shares. If you are a United States person that is an individual, estate or trust, you are encouraged to consult your tax advisors regarding the applicability of this Medicare tax to your income and gains in respect of your investment in our ordinary shares.
Information Reporting and Backup Withholding
U.S. Holders may be required to file certain U.S. information reporting returns with the IRS with respect to an investment in our ordinary shares, including, among others, IRS Form 8938 (Statement of Specified Foreign Financial Assets). As described above under “Passive Foreign Investment Company Consequences”, each U.S. Holder who is a shareholder of a PFIC must file an annual report containing certain information. U.S. Holders paying more than $100,000 for our ordinary shares may be required to file IRS Form 926 (Return by a U.S. Transferor of Property to a Foreign Corporation) reporting this payment. Substantial penalties may be imposed upon a U.S. Holder that fails to comply with the required information reporting.
Dividends on and proceeds from the sale or other disposition of our ordinary shares may be reported to the IRS unless the U.S. Holder establishes a basis for exemption. Backup withholding may apply to amounts subject to reporting if the holder (i) fails to provide an accurate U.S. taxpayer identification number or otherwise establish a basis for exemption, or (ii) is described in certain other categories of persons. However, U.S. Holders that are corporations generally are excluded from these information reporting and backup withholding tax rules.
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules generally will be allowed as a refund or a credit against a U.S. Holder’s U.S. federal income tax liability if the required information is furnished by the U.S. Holder on a timely basis to the IRS.
U.S. Holders should consult their own tax advisors regarding the backup withholding tax and information reporting rules.
YOU ARE URGED TO CONSULT ITS OWN TAX ADVISOR ABOUT THE TAX CONSEQUENCES TO IT OF AN INVESTMENT IN OUR ORDINARY SHARES IN LIGHT OF THE INVESTOR’S OWN CIRCUMSTANCES.
Prospective investors should consult their professional advisers on the possible tax consequences of buying, holding or selling any ordinary shares under the laws of their country of citizenship, residence or domicile.
The following is a discussion on certain Cayman Islands income tax consequences of an investment in the ordinary shares. The discussion is a general summary of present law, which is subject to prospective and retroactive change. It is not intended as tax advice, does not consider any investor’s particular circumstances, and does not consider tax consequences other than those arising under Cayman Islands law.
British Virgin Islands Taxation
There is no withholding tax, capital gains tax, capital transfer tax, estate duty, inheritance tax, succession tax or gift tax in the British Virgin Islands and any dividends, interest, rents, royalties, compensations and other amounts paid by our subsidiary in the British Virgin Islands are exempt from any taxation in the British Virgin Islands imposed under the British Virgin Islands Income Tax Ordinance (Cap 206) provided that they do not relate to real estate in the BVI.
Cayman Islands Taxation
The Cayman Islands currently levies no taxes on individuals or corporations based upon profits, income, gains or appreciation and there is no taxation in the nature of inheritance tax or estate duty. There are no other taxes likely to be material to us levied by the government of the Cayman Islands except for stamp duties which may be applicable on instruments executed in, or brought within the jurisdiction of the Cayman Islands. The Cayman Islands is a party to a double tax treaty entered with the United Kingdom in 2010 but is otherwise not party to any double tax treaties that are applicable to any payments made to or by our company. There are no foreign exchange controls or foreign exchange regulations or currency restrictions in the Cayman Islands.
Payments of dividends and capital in respect of our ordinary shares will not be subject to taxation in the Cayman Islands and no withholding will be required on the payment of a dividend or capital to any holder of our ordinary shares, as the case may be, nor will gains derived from the disposal of our ordinary shares be subject to Cayman Islands income or corporation tax.
The Cayman Islands enacted the International Tax Co-operation (Economic Substance) Act (Revised) together with the Guidance Notes published by the Cayman Islands Tax Information Authority from time to time. The Company is required to comply with the economic substance requirements from July 1, 2019 and make an annual report in the Cayman Islands as to whether or not it is carrying on any relevant activities and if it is, it must satisfy an economic substance test.
63
Singapore Taxation
Individual Income Tax
An individual is a tax resident in Singapore in a year of assessment if, in the preceding year, he resides in Singapore except for such temporary absences as may be reasonable and not inconsistent with a claim by such person to be resident in Singapore. This includes a person who is physically present in Singapore or exercises an employment (other than as a director of a company) in Singapore for 183 days or more during the year preceding the year of assessment.
Generally, individual taxpayers are subject to Singapore income tax on income accruing in or derived from Singapore, unless certain exemptions apply. Foreign-sourced income received in Singapore by a non-resident individual is exempt from Singapore income tax. Foreign-sourced income received on or after January 1, 2004 by a Singapore tax resident individual (except for income received through a partnership in Singapore) is also exempt from Singapore income tax if the Comptroller of Income Tax in Singapore (“Comptroller”) is satisfied that the tax exemption would be beneficial to the individual.
A Singapore tax resident individual is taxed at progressive rates ranging from 0% to 22%. Non-resident individuals, subject to certain exceptions and conditions, are subject to Singapore income tax on income accruing in or derived from Singapore at the rate of 22%.
Corporate Income Tax
A company is regarded as resident in Singapore for Singapore tax purposes if the control and management of its business are exercised in Singapore.
A company is subject to Singapore income tax on income accruing in or derived from Singapore and on foreign-sourced income received or deemed to be received in Singapore, unless certain exemptions apply.
Foreign-sourced income in the form of dividends, branch profits and service income received or deemed to be received in Singapore by a Singapore tax resident company is exempt from Singapore income tax if the following conditions are met:
(i) such income is subject to tax of a similar character to income tax (by whatever name called) under the law of the territory from which such income is received;
(ii) at the time the income is received in Singapore, the highest rate of tax of a similar character to income tax (by whatever name called) levied under the law of the territory from which the income is received on any gains or profits from any trade or business carried on by any company in that territory at that time is not less than 15%; and
(iii) the Comptroller is satisfied that the tax exemption would be beneficial to the Singapore tax resident company.
SingaporeCo incorporated in Singapore was subject to 17% corporate tax rate on its taxable income assessable profits generated from operations arising in or derived from Singapore. From the year of assessment (“YA”) 2020 onwards, three-quarters of a company’s first S$10,000 its normal chargeable income, and half of its next S$190,000 of normal chargeable income are exempt from corporate tax.
Newly incorporated companies will also, subject to certain conditions and exceptions, be eligible for tax exemption on three-quarters of the company’s first S$100,000 of normal chargeable income, and half of its next $100,000 of normal chargeable income, for each of the company’s first three YAs falling in or after YA 2020.
Hong Kong Taxation
HKco and ViraxImmune T cell incorporated in Hong Kong were subject to 16.5% Hong Kong profits tax on their taxable income assessable profits generated from operations arising in or derived from Hong Kong for the years of assessment of 2019/2020 and 2018/2019. As from year of assessment of 2019/2020 onwards, Hong Kong profits tax rates are 8.25% on assessable profits up to HK$2,000,000, and 16.5% on any part of assessable profits over HK$2,000,000. Under Hong Kong tax laws, our Hong Kong subsidiaries are exempted from Hong Kong income profits tax on its foreign- derived income profits. In addition, payments of dividends from our Hong Kong subsidiary to us are not subject to any withholding tax in Hong Kong.
F. Dividends and paying agents.
Not applicable.
G. Statement by experts.
Not applicable.
H. Documents on display.
64
We are subject to the information reporting requirements of the Exchange Act, applicable to foreign private issuers and under those requirements will file reports with the SEC. The SEC maintains an Internet website that contains reports and other information regarding issuers that file electronically with the SEC. You may read and copy this annual report, including the related exhibits and schedules, and any document we file with the SEC at http://www.sec.gov.
As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file annual, quarterly and current reports and financial statements with the SEC as frequently or as promptly as United States companies whose securities are registered under the Exchange Act. However, we file with the SEC, within four months after the end of each fiscal year, or such applicable time as required by the SEC, an annual report on Form 20-F containing financial statements audited by an independent registered public accounting firm, and submit to the SEC, on a Form 6-K, unaudited quarterly financial information for the first three quarters of each fiscal year within 60 days after the end of each such quarter, or such applicable time as required by the SEC.
We maintain a corporate website at www.viraxbiolabs.com. Information contained on, or that can be accessed through, our website does not constitute a part of this Annual Report on Form 20-F. We have included our website address in this Annual Report on Form 20-F solely as an inactive textual reference.
I. Subsidiary Information.
Not applicable.
Item 11. Quantitative and Qualitative Disclosures About Market Risk.
Market risk is the risk of loss related to changes in market prices, including interest rates and foreign exchange rates, of financial instruments that may adversely impact our consolidated financial position, results of operations or cash flows.
Interest Rate Risk
We do not anticipate undertaking any significant long-term borrowings. At present, our investments consist primarily of cash and cash equivalents. We may invest in investment-grade marketable securities with maturities of up to three years, including commercial paper, money market funds, and government/non-government debt securities. The primary objective of our investment activities is to preserve principal while maximizing the income that we receive from our investments without significantly increasing risk and loss. Our investments are exposed to market risk due to fluctuation in interest rates, which may affect our interest income and the fair market value of our investments, if any. We manage this exposure by performing ongoing evaluations of our investments. Due to the short-term maturities, if any, of our investments to date, their carrying value has always approximated their fair value. If we decide to invest in investments other than cash and cash equivalents, it will be our policy to hold such investments to maturity in order to limit our exposure to interest rate fluctuations.
Foreign Currency Exchange Risk
Our foreign currency exposures give rise to market risk associated with exchange rate movements of the U.S dollar, our functional and reporting currency, mainly against the renminbi and the euro. Although the U.S dollar is our functional currency, a portion of our expenses are denominated in both renminbi and euro and currently all of our revenues are denominated in dollars. We do not anticipate that a sizable portion of our expenses will be denominated in currencies other than the U.S dollar. To date, fluctuations in the exchange rates have not materially affected our results of operations or financial condition for the periods under review.
To date, we have not engaged in hedging transactions. In the future, we may enter into currency hedging transactions to decrease the risk of financial exposure from fluctuations in the exchange rates of our principal operating currencies. These measures, however, may not adequately protect us from the material adverse effects of such fluctuations.
Item 12. Description of Securities Other than Equity Securities.
A. Debt Securities.
Not applicable.
B. Warrants and Rights.
Not applicable.
65
C. Other Securities.
Not applicable.
D. American Depositary Shares.
Not applicable
PART II Item 13. Defaults, Dividend Arrearages and Delinquencies.
Not applicable.
Item 14. Material Modifications to the Rights of Security Holders and Use of Proceeds.
See “Item 10. Additional Information” for a description of the rights of shareholders, which remain unchanged.
Use of Proceeds
The following “Use of Proceeds” information relates to the registration statement on Form F-1, as amended (File Number 333-263694) for our initial public offering, which was declared effective by the SEC on June 30, 2022. On July 25, 2022, the Company completed its IPO of 1,350,000 shares of common stock at a price to the public of $5.00 per share for $6.75 million. In addition, on July 25, 2022, Boustead Securities, LLC, as representative of several underwriters, exercised an over-allotment option (the “Option”) in part to purchase 202,500 ordinary shares from the Company in connection with the IPO at a price of $5.00 per ordinary share for $1.01 million. The net proceeds raised from the initial public offering were $6,557,570 after deducting underwriting discounts and the offering expenses payable by us.
The Company filed resale F-1 to register the resale of ordinary shares and ordinary shares issuable upon exercise of the warrants (File Number 333-270734) offered by certain selling shareholders. The Company did not receive any proceeds from the sale of ordinary shares by the selling shareholder. However, the Company received approximately $3.8 million in gross proceeds from the exercise of the warrants as the warrant holders exercised the warrants.
The Company filed resale F-1 to register the resale of ordinary shares and ordinary shares issuable upon exercise of the warrants (File Number 333-268486) offered by certain selling shareholders. The Company did not receive any proceeds from the sale of ordinary shares by the selling shareholder. However, the Company received approximately $4.0 million in gross proceeds from the exercise of the warrants as the warrant holders exercised the warrants.
The Company filed a registration statement on Form F-3 (File Number 333-275893) to register the resale of ordinary shares issuable upon exercise of certain warrants offered by certain selling shareholders and securities. The Company did not receive any proceeds from the sale of ordinary shares by the selling shareholder. However, the Company received $1.9 million in gross proceeds from the exercise of the warrants as the warrant holders exercised the warrants. The Company did not use or receive any proceeds from the At-the-Market Offering at the time of filing, pursuant to an At The Market Offering Agreement (the “Sales Agreement”) with H.C. Wainwright & Co., LLC (the “Sales Agent”), acting as the Company’s sales agent.
As of March 31, 2024, we had used $6,247,016 in operating activities and $1,164,449 in investing activities. The proceeds were primarily used for research and development activities, general and administrative costs and the purchases of lab equipment.
Item 15. Controls and Procedures.
Disclosure controls and procedures
Our management, including our chief executive officer, or CEO, and our chief financial officer, or CFO, are responsible for establishing and maintaining our disclosure controls and procedures (within the meaning of Rule 13a-15(e) of the Exchange Act). These controls and procedures were designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC and that such information is accumulated and communicated to our management, including our CEO and CFO, as appropriate, to allow timely
66
decisions regarding required disclosure. We evaluated these disclosure controls and procedures under the supervision of our CEO and CFO as of March 31, 2024. Based upon that evaluation, our management, including our CEO and CFO, concluded that our disclosure controls and procedures as of March 31, 2024 were not effective.
Management’s annual report on internal control over financial reporting
Disclosure controls and procedures (as defined in Exchange Act Rule 15d-15(e)) are designed with the objective of ensuring that information required to be disclosed in our reports filed under the Exchange Act, such as this report, is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures are also designed with the objective of ensuring that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
Due to its inherent limitations, internal controls over financial reporting may not prevent or detect misstatements. In addition, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The Company has determined that it is too small to effectively assess our internal control over financial reporting as of March 31, 2024, based on the framework for Internal Control-Integrated Framework set forth by The Committee of Sponsoring Organizations of the Treadway Commission (COSO) (2013).
There has been no change in the Company’s internal control over financial reporting during the year ended March 31, 2024 that has materially affected, or is reasonably likely to materially affect, the company’s internal control over financial reporting. Management will continue to monitor and evaluate the effectiveness of our internal controls and procedures over financial reporting on an ongoing basis and are committed to taking further action and implementing additional improvements as necessary.
Attestation Report of Registered Public Accounting Firm
Not applicable.
Changes in internal control over financial reporting
There were no changes in our internal control over financial reporting, other than as described above, that occurred during the year ended March 31, 2024 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 16. [Reserved]
Item 16A. Audit committee financial expert.
Our Board of Directors has determined that Mr. Haight, Mr. Erez and Mr. Norton are audit committee financial experts, as defined by applicable SEC regulations. Each audit committee member qualified as an “independent director,” as that term is defined under Nasdaq rules.
Item 16B. Code of Ethics.
We have adopted a code of ethics, referred to as a Code of Business Conduct, applicable to our directors, officers and all other employees. Our code of ethics is publicly available on our website at www.canfite.com. If we make any amendment to the code of ethics or grant any waivers, including any implicit waiver, from a provision of the code of ethics, which applies to our chief executive officer, chief financial officer, chief accounting officer or controller, or persons performing similar functions, we will disclose the nature of such amendment or waiver on our website.
Item 16C. Principal Accountant Fees and Services.
The following table sets forth, for each of the years indicated, the fees billed by our independent registered public accounting firm.
67
|
|
For the Year Ended March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Services Rendered |
|
|
|
|
|
|
||
Audit |
|
$ |
70,000 |
|
|
$ |
52,500 |
|
Audit related services |
|
|
— |
|
|
|
12,500 |
|
Tax |
|
|
— |
|
|
|
— |
|
All other fees |
|
|
18,500 |
|
|
|
— |
|
Total |
|
$ |
88,500 |
|
|
$ |
65,000 |
|
Audit Committee Pre-Approval Policies and Procedures
Our audit committee’s specific responsibilities in carrying out its oversight of the quality and integrity of the accounting, auditing and reporting practices of us include the approval of audit and non-audit services to be provided by the external auditor. The audit committee approves in advance the particular services or categories of services to be provided to us during the following yearly period and also sets forth a specific budget for such audit and non-audit services. Additional non-audit services may be pre-approved by the audit committee.
Item 16D. Exemptions from the Listing Standards for Audit Committees.
Not applicable.
Item 16E Purchases of Equity Securities by the Issuer and Affiliated Purchasers.
Not applicable.
Item 16F. Change in Registrant’s Certifying Accountant.
On April 19, 2023, Reliant CPA PC replaced BF Borgers CPA P.C. as our independent registered public accounting firm. We previously disclosed this change in our certifying accountant in a current report on Form 6-K filed with the U.S. Securities and Exchange Commission on April 21, 2023.
Item 16G. Corporate Governance.
We are a foreign private issuer whose ordinary shares are listed on the Nasdaq. As such, we are required to comply with U.S. federal securities laws, including the Sarbanes-Oxley Act, and the Nasdaq rules, including the Nasdaq corporate governance requirements. The Nasdaq rules provide that foreign private issuers may follow home country practice in lieu of certain qualitative listing requirements subject to certain exceptions and except to the extent that such exemptions would be contrary to U.S. federal securities laws, so long as the foreign issuer discloses that it does not follow such listing requirement and describes the home country practice followed in its reports filed with the SEC.
Item 16H. Mine Safety Disclosure.
Not applicable.
Item 16I. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
ITEM 16J. Insider Trading Policy
The Company has
68
ITEM 16K. Cybersecurity
We recognize the importance of assessing, identifying and managing material risks associated with cybersecurity threats. These risks include, among other things: operational risks, intellectual property theft, fraud, extortion, harm to employees or customers and violation of data privacy and security laws.
We maintain various cybersecurity measures and protocols to safeguard our systems and data and continuously monitor and assess potential threats to pre-emptively address any emerging cyber risks. We have implemented various processes for assessing, identifying, and managing material risks from cybersecurity threats, which are integrated into our overall risk management framework. These processes include access controls to organizational systems, data encryption, cybersecurity training and security awareness campaigns through direct mail, and are designed to systematically evaluate potential vulnerabilities and cybersecurity threats and minimize their potential impact on our organization’s operations, assets, and stakeholders. Our cybersecurity risk management processes share common methodologies, reporting channels and governance processes with our broader risk management processes. By embedding cybersecurity risk management into and aligning it with our broader risk management processes, we aim to ensure a comprehensive and proactive approach to safeguarding our assets and operations.
We engage assessors, consultants, auditors, and other third-party specialists to enhance the effectiveness of our cybersecurity processes, augment our internal capabilities, validate our controls, and stay abreast of evolving cybersecurity risks and best practices.
For the fiscal year ended March 31, 2024, we did not detect any cybersecurity incidents that have materially affected or are reasonably likely to materially affect us, including our business strategy, results of operations, or financial condition.
Responsibility for overseeing cybersecurity risks is integrated into our internal cybersecurity committee (the "Cybersecurity Committee"), which includes our Chief Technical Officer, Chief Financial Officer and other key executives. We also utilize third-party service providers who are responsible for monitoring, detecting and assessing cybersecurity risks and incidents. These third-party service providers are also used for certain IT-related services, where appropriate, to assess, test or otherwise assist with aspects of our security controls. Accordingly, we also implement processes to oversee and identify material cybersecurity risks associated with our utilization of third-party service providers on whom we have a material dependency, such as conducting due diligence assessments to evaluate their cybersecurity measures, data protection practices, and compliance with relevant regulatory requirements.
Our third-party service providers currently comprises senior IT professionals with expertise in risk management, cybersecurity, and information technology. These individuals have, and any future individuals are expected to have credentials relevant to their role, which includes prior experience working in similar roles and formal education. The third-party service providers are also expected to keep abreast of cybersecurity best practices and procedures. The third-party service providers are responsible for assessing, identifying and mitigating material cybersecurity risks, including at a strategic level, monitoring for, defending against and remediating cybersecurity incidents and implementing and making improvements to our overall cybersecurity strategy.
As we do not have a dedicated board committee solely focused on cybersecurity, our full board oversees the implementation of our cybersecurity strategy, as well as cybersecurity risks, with the aim of protecting our interests and assets. Our cybersecurity strategy was developed by Cybersecurity Committee, the third-party service providers and approved by senior management. The board receives periodic reports and presentations on cybersecurity risks from the Cybersecurity Committee, including incidents or breaches (if any), vulnerabilities, mitigation strategies and the overall effectiveness of our cybersecurity program. These reports highlight significant or emerging cybersecurity threats, their potential impact on the organization, ongoing initiatives to mitigate risks and any proposed actions or investments required to enhance our cybersecurity posture.
PART III
Item 17. Financial Statements.
We have responded to Item 18 in lieu of responding to this item.
Item 18. Financial Statements.
Please refer to the financial statements beginning on page F-1.
Item 19. Exhibits.
EXHIBIT INDEX
69
Exhibit |
|
Description of Exhibit |
1.1 |
|
|
2.1 |
|
|
2.2 |
|
|
2.3 |
|
|
4.1 |
|
|
4.2 |
|
|
4.3 |
|
|
4.4 |
|
|
4.5 |
|
|
4.6 |
|
|
8.1 |
|
|
10.1 |
|
|
10.2 |
|
|
10.3 |
|
|
10.4 |
|
|
10.5 |
|
|
10.6 |
|
|
10.7 |
|
|
10.8 |
|
|
10.9 |
|
|
11.1 |
|
|
12.1** |
|
CEO Certification Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
12.2** |
|
CFO Certification Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
13.1** |
|
CEO Certification Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
13.2** |
|
CFO Certification Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
70
97.1 |
|
|
99.1 |
|
|
99.2 |
|
* |
Filed herewith |
** |
Furnished herewith. |
71
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this Annual Report on Form 20-F on its behalf.
|
Virax Biolabs Group Limited |
|
|
/s/ James Foster |
|
August 20, 2024 |
Chief Executive Officer |
|
72
Virax Biolabs Group Limited
Index to Consolidated Financial Statements
|
|
Page |
Consolidated Financial Statements for the Years Ended March 31, 2024, 2023 and 2022 |
|
|
Report of Independent Registered Public Accounting Firm (PCAOB ID Number |
|
F-2 |
|
|
|
Financial Statements: |
|
|
Consolidated Statements of Financial Position as of March 31, 2024, and 2023 |
|
F-3 |
|
F-4 |
|
|
F-5 |
|
Consolidated Statements of Cash Flows for the Years Ended March 31, 2024, 2023 and 2022 |
|
F-6 |
Notes to Consolidated Financial Statements for the Years Ended March 31, 2024, 2023 and 2022 |
|
F-7 |
F-1

Report of Independent Registered Public Accounting Firm
To the shareholders and the board of directors of Virax Biolabs Group Limited
Opinion on the Financial Statements
We have audited the accompanying consolidated statements of financial position of Virax Biolabs Group Limited (the “Company”), as of March 31, 2024 and 2023, the related consolidated statements of loss and comprehensive loss, changes in stockholders’ equity and cash flows for each of the three years in the period ended March 31, 2024, and related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of March 31, 2024 and 2023, and the results of its operations and its cash flows for each of the three years in the period ended March 31, 2024, in conformity with the International Financial Reporting Standards as issued by the International Accounting Standards Board.
Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company’s significant operating losses raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
/s/ Reliant CPA PC
Served as Auditor since 2023
August 20, 2024
F-2
VIRAX BIOLABS GROUP LIMITED
CONSOLIDATED STATEMENTS OF FINANCIAL POSITION
|
|
As of March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Assets |
|
|
|
|
|
|
||
Current assets: |
|
|
|
|
|
|
||
Cash |
|
$ |
|
|
$ |
|
||
VAT receivable |
|
|
|
|
|
— |
|
|
Inventories |
|
|
|
|
|
— |
|
|
Prepaid expenses and deposits |
|
|
|
|
|
|
||
Total current assets |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Non-current assets |
|
|
|
|
|
|
||
Right-of-use assets, net |
|
|
|
|
|
— |
|
|
Property, plant & equipment, net |
|
|
|
|
|
— |
|
|
Intangible software, net |
|
|
— |
|
|
|
|
|
Total non-current assets |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Total assets |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
Liabilities |
|
|
|
|
|
|
||
Current liabilities: |
|
|
|
|
|
|
||
Accounts payable and accrued liabilities |
|
$ |
|
|
$ |
|
||
Current operating lease liabilities |
|
|
|
|
|
— |
|
|
Accounts payable - related parties |
|
|
— |
|
|
|
|
|
Note payable |
|
|
— |
|
|
|
|
|
Deferred revenue |
|
|
— |
|
|
|
|
|
Total current liabilities |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Non-current liabilities |
|
|
|
|
|
|
||
Non-current operating lease liabilities |
|
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
||
Total liabilities |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
||
Stockholders’ equity |
|
|
|
|
|
|
||
Ordinary shares, $ |
|
|
|
|
|
|
||
Reserves |
|
|
|
|
|
|
||
Accumulated deficit |
|
|
( |
) |
|
|
( |
) |
Accumulated other comprehensive income (loss ) |
|
|
|
|
|
( |
) |
|
Total stockholders’ equity (Virax) |
|
|
|
|
|
|
||
Non-Controlling Interest |
|
|
( |
) |
|
|
( |
) |
Total stockholders’ equity |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Total liabilities and stockholders’ equity |
|
$ |
|
|
$ |
|
||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
VIRAX BIOLABS GROUP LIMITED
CONSOLIDATED STATEMENTS OF LOSS
AND OTHER COMPREHENSIVE LOSS
|
|
For the Year Ended March 31, |
|
|||||||||
|
|
2024 |
|
|
2023 |
|
|
2022 |
|
|||
Revenue |
|
$ |
|
|
$ |
|
|
$ |
— |
|
||
Cost of revenue |
|
|
|
|
|
|
|
|
— |
|
||
Gross profit |
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Operating expenses: |
|
|
|
|
|
|
|
|
|
|||
General and administrative |
|
|
|
|
|
|
|
|
|
|||
Research & development |
|
|
|
|
|
|
|
|
|
|||
Impairment of intangible asset |
|
|
|
|
|
— |
|
|
|
— |
|
|
Total operating expenses |
|
$ |
|
|
$ |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
Operating loss |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
Other income (expenses): |
|
|
|
|
|
|
|
|
|
|||
Interest expense, net |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Other income (expense), net |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
Total other income (expenses), net |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
Loss before income taxes |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
Income tax expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|||
Net loss |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
Net loss attributable to non-controlling interest |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Net loss attributable to Virax shareholders |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
Other comprehensive loss: |
|
|
|
|
|
|
|
|
|
|||
Foreign currency adjustment |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Comprehensive loss |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
Comprehensive loss attributable to non-controlling interest |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Comprehensive loss attributable to Virax |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
Basic and diluted weighted average shares outstanding |
|
|
|
|
|
|
|
|
|
|||
Ordinary shares |
|
|
|
|
|
|
|
|
|
|||
Basic and diluted net loss per share to Virax shareholders |
|
|
|
|
|
|
|
|
|
|||
Ordinary shares |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
The accompanying notes are an integral part of these consolidated financial statements.
F-4
VIRAX BIOLABS GROUP LIMITED
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS' EQUITY
|
|
Ordinary shares |
|
|
Reserves |
|
|
Subscription Receivable |
|
|
Accumulated Deficit |
|
|
Accumulated |
|
|
Total Stockholders’ Equity |
|
|
Non |
|
|
Total Stockholders’ Equity |
|
||||||||||||
|
|
Shares |
|
|
Amount |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
Balance at March 31, 2021 |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|||
Shares issued for cash |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||||
Shares issued for services |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||||
Shares issued for settlement of related-party payables |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||||
Shares issued for conversion of convertible debt |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||||
Surrender of ViraxClear Shares |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Imputed interest |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
Foreign currency adjustment |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Net Loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Balance at March 31, 2022 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|||
Shares issued for cash |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||||
Shares issued for services |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||||
Shares issued for settlement of related-party payables |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||||
Contributed capital - settlement of related party debt |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
Cashless warrant exercise |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
||||||
Pre-funded warrant exercise |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
Foreign currency adjustment |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
Net Loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Balance at March 31, 2023 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||||
Pre-funded warrant exercise |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
Warrant exercise |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||||
Shares issued for settlement of debt |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||||
Shares issued for rounding on Share Consolidation |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
Foreign currency adjustment |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
Net Loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Balance at March 31, 2024 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||||
All share amounts have been given retroactive effect in the financial statements as a result of the Share Restructuring on June 19, 2022 and the Share Consolidation on December 18, 2023. See Note 18.
The accompanying notes are an integral part of these consolidated financial statements.
F-5
VIRAX BIOLABS GROUP LIMITED
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
|
For the years ended |
|
|||||||||
|
|
2024 |
|
|
2023 |
|
|
2022 |
|
|||
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
|||
Depreciation and amortization |
|
|
|
|
|
— |
|
|
|
— |
|
|
Amortization of right-to-use assets |
|
|
|
|
|
— |
|
|
|
— |
|
|
Impairment of intangible asset |
|
|
|
|
|
— |
|
|
|
— |
|
|
Accretion expense |
|
|
|
|
|
— |
|
|
|
— |
|
|
Stock-based compensation |
|
|
|
|
|
|
|
|
— |
|
||
Stock issued for services |
|
|
— |
|
|
|
|
|
|
|
||
Gain on debt extinguishment |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Other expense |
|
|
|
|
|
— |
|
|
|
|
||
Foreign currency translation (gain) loss |
|
|
|
|
|
|
|
|
|
|||
Net changes in operating assets & liabilities: |
|
|
|
|
|
|
|
|
|
|||
Accounts receivable |
|
|
— |
|
|
|
— |
|
|
|
|
|
Prepaid expenses and deposits |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
Inventories |
|
|
( |
) |
|
|
|
|
|
|
||
VAT receivable |
|
|
( |
) |
|
|
— |
|
|
|
— |
|
Deferred revenue |
|
|
( |
) |
|
|
|
|
|
— |
|
|
Accounts payable and accrued liabilities |
|
|
( |
) |
|
|
|
|
|
|
||
Net cash used in operating activities |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|||
Investment - internally developed software |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
Purchase of property, plant & equipment |
|
|
( |
) |
|
|
— |
|
|
|
— |
|
Net cash used in investing activities |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
— |
|
|
|
|
|
|
|
|
|
|
|
|||
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|||
Lease payments |
|
|
( |
) |
|
|
|
|
|
|
||
Proceeds from (payments to) related parties |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
Proceeds from shares issuance for cash |
|
|
|
|
|
|
|
|
|
|||
Proceeds from warrant exercise |
|
|
|
|
|
— |
|
|
|
— |
|
|
Proceeds from note payable |
|
|
— |
|
|
|
|
|
|
|
||
Payments on note payable |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
Net cash provided by financing activities |
|
$ |
|
|
$ |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
Net change in cash |
|
|
( |
) |
|
|
|
|
|
|
||
Cash at beginning of year |
|
|
|
|
|
|
|
|
|
|||
Cash at end of year |
|
$ |
|
|
$ |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
Supplemental disclosure of cash flow information |
|
|
|
|
|
|
|
|
|
|||
Cash paid during the year for: |
|
|
|
|
|
|
|
|
|
|||
Interest |
|
$ |
|
|
$ |
|
|
$ |
— |
|
||
Income taxes |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|||
Supplemental disclosure of non-cash investing and financing activities: |
|
|
|
|
|
|
|
|
|
|||
Addition of Right-of-use assets |
|
|
|
|
|
— |
|
|
|
— |
|
|
Shares issued for conversion of convertible debt |
|
|
— |
|
|
|
— |
|
|
|
|
|
Shares issued for settlement of related party payable |
|
|
— |
|
|
|
|
|
|
|
||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
VIRAX BIOLABS GROUP LIMITED
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED MARCH 31, 2023 AND 2022
Note 1 — General information and reorganization transactions
Virax Biolabs is a holding company incorporated as an exempted company under the laws of the Cayman Islands. As a holding company with no material operations of its own, it conducts the majority of its operations through its subsidiaries in the United Kingdom, the United States, and China and has been operating since 2013.
The Company is a global innovative biotechnology company focused on the prevention, detection, diagnosis, and risk management of viral diseases with a current focus on the field of T cell in Vitro Diagnostics. The Company is in the process of developing and manufacturing tests that can predict adaptive immunity to viral diseases as well as identify individuals suffering from T cell exhaustion and chronic inflammation linked to post viral syndromes. The Company's mission is to protect people from viral diseases and help with the early diagnosis of post viral syndromes associated with T cell exhaustion and chronic inflammation through the provision of diagnostic tests and tests for adaptive immunity.
The Company is in the process of developing ViraxImmune, with the intention of providing an immunology profiling platform that assesses each individual’s immune risk profile against major global viral diseases as well as helping with the early diagnosis of post viral syndromes associated with T cell exhaustion and chronic inflammation. We are in the process of developing a T cell Test under the ViraxImmune brand and will apply for regulatory agency approval in the future. We believe that the T cell Tests and immunology platform we are developing under the ViraxImmune brand will be particularly useful in assisting in the threat analysis of the major viruses faced globally as well as helping with the early diagnosis of indications associated with T cell exhaustion and chronic inflammation which have been linked to symptoms such as chronic fatigue. Initially, we will be focusing on diseases associated with post viral syndromes including but not limited to SARS-CoV-2, Human Papillomavirus (better known as HPV), Malaria, Hepatitis B, and Herpes (better known as HSV-1).
The Company also has the ability to distribute existing diagnostics test kits through our ViraxClear and ViraxVet brands. Currently, we do not manufacture or develop any product that we sell in our ViraxClear and ViraxVet product portfolios, and we act as a distributor of third-party suppliers’ products. While ViraxClear and ViraxVet have the ability to sell and distribute existing test kits, they are not the main focus of the Company.
Virax Biolabs Group Limited (“Virax Biolabs”) — Virax Biolabs Group Limited is a Cayman Islands exempted company incorporated on
Virax Biolabs (UK) Limited (“Virax UK”) — Virax Biolabs (UK) Limited was incorporated on
Virax Biolabs Limited (“HKCo” or formerly known as Shanghai Biotechnology Devices Ltd.) — Virax Biolabs Limited, incorporated on
Virax Immune T- Cell Medical Device Company Limited (“Virax Immune T cell”) — Virax Immune T cell Medical Device Company Limited, a wholly-owned subsidiary of HKCo, incorporated on
Virax Biolabs Pte. Limited (“SingaporeCo”) — Virax Biolabs Pte. Limited, incorporated on
Logico Bioproducts Corp. (“Logico BVI”) — Logico Bioproducts Corp., a wholly-owned subsidiary of SingaporeCo, is a limited liability company incorporated in the British Virgin Islands on
Shanghai Xitu Consulting Co., Limited (“Shanghai Xitu”) — Shanghai Xitu, a wholly-owned subsidiary of Logico BVI and a wholly foreign owned enterprise, is a limited liability company incorporated on
F-7
Virax Biolabs USA Management, Inc. — Virax Biolabs USA Management, Inc. was incorporated on
Virax Biolabs Group Holdings Ltd (“Virax UK HoldCo”) — Virax Biolabs Group Holdings Limited was incorporated on
Virax Biolabs FZ-LLC (“Virax Dubai”) — Virax Biolabs FZ-LLC was incorporated on
Virax Biolabs Trading B.V. (“Virax Netherlands”) — Virax Biolabs Trading B.V. was incorporated on
Virax Biolabs UK Operating Limited (“Virax UK Operating”) — Virax Biolabs UK Operating Limited was incorporated on
These financial statements are presented in US dollars.
Going Concern
The Company has recurring losses, accumulated deficit totaling $
Management plans to fund its cash flow needs through current cash on hand and future debt and/or equity financings which it may obtain through one or more public or private equity offerings, debt financings, government or other third-party funding, strategic alliances, or collaboration agreements.
The accompanying financial statements have been prepared on a going concern basis, which assumes that the Company will be able to continue in operation for the foreseeable future and will be able to realize its assets and discharge its liabilities and commitments in the normal course of business. Until such time that the Company implements its growth strategy, it expects to continue to generate operating losses in the foreseeable future, mostly due to research and development activities, corporate overhead and costs of being a public company. The Company will need to generate additional revenue or raise additional capital in the near term to fund its ongoing operations and business activities.
Note 2 — Summary of Significant Accounting Policies
This summary provides a list of the significant accounting policies adopted in the preparation of these consolidated financial statements to the extent they have not been disclosed in the other notes below. The policies have been consistently applied to all the years presented, unless otherwise stated.
Basis of preparation
Compliance with IFRS
The consolidated financial statements of the Company have been prepared on a going concern basis and in accordance with International Financial Reporting Standards (“IFRS”) and interpretations issued by the IFRS Interpretations Committee (“IFRS IC”) applicable to companies reporting under IFRS. The financial statements comply with IFRS as issued by the International Accounting Standards Board (“IASB”).
Certain prior year amounts have been reclassified for consistency with the current year presentation. These reclassifications had no effect on the reported results of operations. For the full year ended March 31, 2023, the Company has reclassified certain general and administrative expenses to research and development expenses.
F-8
Historical cost convention
The consolidated financial statements have been prepared on a historical cost basis, as modified by the revaluation of certain financial assets and liabilities which are recognized at fair value through consolidated statements of profit and loss and other comprehensive loss.
Principles of consolidation
Subsidiaries are all entities over which the Company has control. The Company controls an entity when the Company is exposed to, or has rights to, variable returns from its involvement with the entity and has the ability to affect those returns through its power to direct the activities of the entity. Subsidiaries are fully consolidated from the date on which control is transferred to the Company. They are deconsolidated from the date that control ceases.
The following table lists the constituent companies in the Company.
Company names |
|
Jurisdiction |
|
Incorporation |
|
Ownership |
Virax Biolabs Group Limited |
|
|
|
Holding Company |
||
Virax Biolabs (UK) Limited |
|
|
|
|||
Virax Biolabs Limited |
|
|
|
|||
Virax Immune T cell Medical Device Company Limited |
|
|
|
|||
Virax Biolabs PTE. Limited |
|
|
|
95.54% (via Virax Biolabs Limited) |
||
Logico Bioproducts Corp. |
|
|
|
|||
Shanghai Xitu Consulting Co., Ltd |
|
|
|
|||
Virax Biolabs USA management, Inc. |
|
|
|
|||
Virax Biolabs Group Holdings Ltd |
|
|
|
|||
Virax Biolabs FZ-LLC |
|
|
|
|||
Virax Biolabs Trading B.V. |
|
|
|
|||
Virax Biolabs UK Operating Limited |
|
|
|
Inter-company transactions, balances and unrealized gains on transactions between the subsidiaries are eliminated. Unrealized losses are also eliminated unless the transaction provides evidence of an impairment of the transferred asset. Accounting policies of subsidiaries have been changed where necessary to ensure consistency with the policies adopted by the Company.
Common Control and Share Restructuring
Virax Cayman was formed on September 2, 2021. As all the Company's subsidiaries presented are under common control, the series of contractual arrangements between SingaporeCo, HKCo, Virax UK, and the Company during the year ended March 31, 2022 constituted a reorganization under common control and were required to be retrospectively applied to the consolidated financial statements at their historical amounts. The Company has a non-controlling interest due to certain shareholders of SingaporeCo, representing a minority interest of approximately 4.46%, not ultimately converting their interest in the subsidiaries. The consolidated financial statements for the year ended March 31, 2022 have been prepared as if the existing corporate structure had been in existence throughout all periods
F-9
ended March 31, 2022. This includes a retrospective presentation to reflect the effects of the reorganization in accordance with IFRS as of March 31, 2021. See the table below:
|
SingaporeCo Shares as of March 31, 2021 |
HKCo Shares Issued for 95.54% of SingaporeCo on June 24, 2021 |
HKCo issued shares after the stock split as of April 30, 2021 |
HKCo issued shares after June 24, 2021 |
HKCo issued shares as of September 20, 2021 |
Number of shares issued per share exchange agreement as of September 30, 2021 (pre-share consolidation) |
Class A |
178,048,513 |
19,111,119 |
80,000,000 |
3,367,409 |
102,478,548 |
2,556,555 |
Class B |
|
|
|
|
|
7,026,759 |
|
|
|
|
|
|
9,583,314 |
Segment information
The Company has
Foreign currency translation
Functional and presentation currency
Items included in the financial statements of each of the Company’s entities are measured using the currency of the primary economic environment in which the entity operates (the “functional currency”). The consolidated financial statements are presented in US dollar, which is the Company’s presentation currency.
Entity |
|
Functional Currency |
Virax Biolabs Group Limited |
|
|
Virax Biolabs (UK) Limited |
|
|
Virax Biolabs Limited |
|
|
ViraxImmune T cell Medical Device Company Limited |
|
|
Virax Biolabs PTE. LTD |
|
|
Logico Bioproducts Corp. |
|
|
Shanghai Xitu Consulting Co., Ltd |
|
|
Virax Biolabs USA Management, Inc. |
|
|
Virax Biolabs Group Holdings Ltd |
|
|
Virax Biolabs FZ-LLC |
|
|
Virax Biolabs Trading B.V. |
|
|
Virax Biolabs UK Operating LLC |
|
The results and financial position of foreign operations (none of which has the currency of a hyperinflationary economy) that have a functional currency different from the presentational currency are translated into the presentational currency as follows:
Transactions and balances
Foreign currency transactions are translated into the functional currency using the exchange rates at the dates of the transactions. Foreign exchange gains and losses resulting from the settlement of such transactions and from the translation of monetary assets and liabilities
F-10
denominated in foreign currencies at year-end exchange rates are generally recognized in statements of profit and loss and other comprehensive loss.
Exchange rates
The most important exchange rates per USD 1.00 that have been used in preparing the financial statements are:
|
|
Closing rate |
|
|
Average rate |
|
||||||||||||||||||
|
|
As of March 31, |
|
|
|
|
|
As of March 31, |
|
|||||||||||||||
|
|
2024 |
|
|
2023 |
|
|
2022 |
|
|
2024 |
|
|
2023 |
|
|
2022 |
|
||||||
British Pound |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
Euro |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
Singapore Dollar |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
Renminbi |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
Revenue recognition
Revenues are generally recognized upon the transfer of control of promised products or services provided to the Company’s customers, reflecting the amount of consideration we expect to receive for those products or services. The Company enters into contracts that can include various combinations of products and services. Revenue is recognized net of any taxes collected from customers, which are subsequently remitted to governmental authorities. The revenue recognition policy is consistent for sales generated directly with customers and sales generated indirectly through solution partners and resellers.
Revenues are recognized upon the application of the following steps:
The timing of revenue recognition may differ from the timing of billing our customers. The Company receives payments from customers based on a billing schedule as established in our contracts. Contract assets are recognized when performance is completed in advance of scheduled billings. Deferred revenue is recognized when billings are in advance of performance under the contract. The Company’s revenue arrangements include standard warranty provisions that our products and services will perform and operate in all material respects with the applicable published specifications, the financial impacts of which have historically been, and are expected to continue to be insignificant. Our contracts do not include a significant financing component.
The Company’s products are generally sold without a right of return, so there is no variable consideration when determining the amount of revenue to recognize. Returns and credits are estimated at contract inception and updated at the end of each reporting period if additional information becomes available.
Employee benefits
Share-based payments
The Company accounts for share-based compensation in accordance with IFRS 2 “Share-based payment” (“IFRS 2”), which requires companies to estimate the fair value of equity-based payment awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as an expense over the requisite service periods in the Company’s consolidated statement of profit and loss and other comprehensive loss, based on acceleration method in twelve month tranches.
The Company recognizes compensation expenses for the value of its awards granted based on the vesting attribution approach over the requisite service period of each of the awards, net of estimated forfeitures. IFRS 2 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
The Company estimates the fair value of share options granted using the black-scholes option pricing model. The option-pricing model requires a number of assumptions, of which the most significant are the expected stock price volatility and the expected option term. Expected volatility was calculated based upon historical volatility of the Company. The risk-free interest rate is based on the yield from U.S. treasury bonds with an equivalent term. The Company has historically not paid dividends and has no foreseeable plans to pay dividends.
Warrants
F-11
The Company determines the accounting classification of warrants that are issued, as either liability or equity, by first assessing whether the warrants meet liability classification in accordance with IFRS 9, Financial Instruments, Under IFRS 9, warrants are considered liability-classified if the warrants are mandatorily redeemable, obligate the issuer to settle the warrants or the underlying shares by paying cash or other assets, or must or may require settlement by issuing a variable number of shares.
If the warrants do not meet liability classification, the Company assesses the requirements that contracts that require or may require the issuer to settle the contract for cash are liabilities recorded at fair value, irrespective of the likelihood of the transaction occurring that triggers the net cash settlement feature. If the warrants do not require liability classification, in order to conclude equity classification, the Company assesses whether the warrants are indexed to its common stock and whether the warrants are classified as equity. After all relevant assessments are made, the Company concludes whether the warrants are classified as liability or equity. Liability-classified warrants are required to be accounted for at fair value both on the date of issuance and on subsequent accounting period ending dates, with all changes in fair value after the issuance date recorded as a component of other income (expense), net in the statements of operations. Equity-classified warrants are accounted for at fair value on the issuance date with no changes in fair value recognized after the issuance date. As of March 31, 2024, all of the Company’s outstanding warrants are equity-classified warrants. See Note 18.
Income tax
The income tax expense or credit for the period is the tax payable on the current period’s taxable income based on the applicable income tax rate for each jurisdiction adjusted by changes in deferred tax assets and liabilities attributable to temporary differences and to unused tax losses.
The current income tax expense or credit is calculated on the basis of the tax laws enacted or substantively enacted at the end of the reporting period in the countries where the Company and its subsidiaries operate and generate taxable income. Although the Company is organized as a Cayman Islands corporation, the Company could be subject to income and other taxes in various other jurisdictions, including the United Kingdom, United States, China, Hong Kong and Singapore. Management periodically evaluates positions taken in tax returns with respect to situations in which applicable tax regulation is subject to interpretation. It establishes provisions where appropriate on the basis of amounts expected to be paid to (or recovered from) the tax authorities.
Deferred income tax is provided in full, using the liability method, on temporary differences arising between the tax bases of assets and liabilities and their carrying amounts in the consolidated financial statements. Deferred income tax is determined using tax rates (and laws) that have been enacted or substantively enacted by the end of the reporting period and are expected to apply when the related deferred income tax asset is realized or the deferred income tax liability is settled.
Deferred tax assets are recognized only if it is probable that future taxable profit will be available to utilize those temporary differences and losses.
Deferred tax assets and liabilities are offset when there is a legally enforceable right to offset current tax assets and liabilities and when the deferred tax balances relate to the same taxation authority.
Current and deferred tax is recognized in profit or loss, except to the extent that it relates to items recognized in other comprehensive loss, in which case the tax is also recognized in other comprehensive loss.
Property plant and equipment
Property and equipment are recorded at cost less accumulated depreciation. Expenditures that extend the life of the asset are capitalized and depreciated. Depreciation is recorded using the straight-line method over the estimated useful lives of the assets. Management evaluates the useful lives and method of depreciation at least annually and accounts for any changes to the useful life or method prospectively. Maintenance and repairs are charged to expense as incurred; cost of major additions and betterments are capitalized. Capitalized software is recorded at cost less accumulated amortization and the amortization is recorded using the straight-line method over the estimated useful life of the software.
The estimated useful lives are:
Laboratory equipment |
|
Computer equipment |
|
Capitalized software |
Impairment of assets
Assets are tested for impairment whenever events or changes in circumstances indicate that the carrying amount may not be recoverable. An impairment loss is recognized in profit or loss for the amount by which the asset’s carrying amount exceeds its recoverable amount. The recoverable amount is the higher of an asset’s fair value less costs of disposal and value in use and is calculated with reference to
F-12
future discounted cash flows that the asset is expected to generate when considered as part of a cash-generating unit. Assets that suffered an impairment are reviewed for possible reversal of the impairment at the end of each reporting period. If an impairment subsequently reverses, the carrying amount of the asset is increased to the revised estimate of its recoverable amount, but so that the increased carrying amount does not exceed the carrying amount that would have been determined had no impairment charge been recognized for the asset in prior years.
Leases
The Company accounts for leases under IFRS 16 'Leases'. IFRS 16 introduced a single lease accounting model, requiring a lessee to recognize assets and liabilities for all leases with a term of more than twelve months, unless the underlying asset is of low value. The lessee is required to recognize a right-of-use asset representing the right to use the underlying asset, and a lease liability representing the obligation to pay lease payments.
At the inception of a contract, we assess whether a contract is, or contains a lease by determining whether the contract conveys the right to control the use of an identified asset for a period of time in exchange for consideration. To assess whether a contract conveys the right to control the use of an identified asset, we assess whether:
A right-of-use asset and corresponding lease liability are recognized on the lease commencement date. The right-of-use asset is initially measured at cost, which comprises the initial amount of the lease liability adjusted for any lease payments made at or before the commencement date, plus any initial direct costs incurred and an estimate of costs to dismantle and remove the underlying asset or to restore the underlying asset or the site on which it is located, less any lease incentives received. The right-of-use asset is subsequently amortized using the straight-line method from the commencement date to the end of the lease term. In addition, the right-of-use asset is reduced by impairment losses and adjusted for certain remeasurement of the lease liabilities, if any.
The lease liability is initially measured at the present value of the lease payments that are not paid at the commencement date. The lease payments are discounted using the implicit interest rate in the lease. If the rate cannot be readily determined, our incremental rate of borrowing is used. The lease liability is subsequently measured at amortized cost using the effective interest method. The lease liability is remeasured when there is a change in future lease payments arising from a change in an index or rate, if there is a change in our estimate of the amount expected to be payable under a residual value guarantee, if we change our assessment of whether we will exercise a purchase, extension or termination option, or if the underlying lease contract is amended.
We have elected not to separate fixed non-lease components from lease components and instead account for each lease component and associated fixed non-lease components as a single lease component.
We have elected not to recognize right-of-use assets and lease liabilities for short-term leases that have a lease term of 12 months or less. We recognize the lease payments associated with these leases as an expense on a straight-line basis over the lease term.
Inventories
Inventories are stated at the lower of cost and net realizable value. Cost is determined using the first-in, first-out (FIFO) method. The cost of finished goods comprises cost of purchase and, where appropriate, other directly attributable costs. It excludes borrowing costs. Net realizable value is the estimated selling price in the ordinary course of business less estimated costs necessary to make the sale.
Accounts receivable
Accounts receivables are amounts due from customers for goods sold or services performed in the ordinary course of business. Trade receivables are recognized initially at fair value. The Company holds trade receivables with the objective to collect the contractual cash flows and therefore measures them subsequently at amortized cost using the effective interest method, less provision for impairment. If collection is expected in one year or less, they are classified as current assets. If not, they are presented as non-current assets.
Cash
For the purposes of presentation in the consolidated statements of cash flows, cash includes cash in hand.
F-13
Share capital and reserves
Ordinary shares are classified as equity. Incremental costs directly attributable to the issue of new shares are shown in equity as a deduction from the proceeds of the issue.
Accounts payables and accrued liabilities
Accounts payable and accrued liabilities are liabilities for goods and services provided to the Company prior to the end of the financial year which are unpaid. They are recognized initially at their fair value and subsequently measured at amortized cost using the effective interest method. They are classified as current liabilities if payment is due within one year or less. If not, they are presented as non-current liabilities. All the accounts payable and accrued liabilities were current for the years ended March 31, 2024 and 2023.
Financial Instruments
Classification
The Company classifies its financial instruments in the following categories: at fair value through profit and loss (“FVTPL”), at fair value through other comprehensive income (loss) (“FVTOCI”) or at amortized cost. The Company determines the classification of financial assets at initial recognition. The classification of debt instruments is driven by the Company’s business model for managing the financial assets and their contractual cash flow characteristics. Equity instruments that are held for trading are classified as FVTPL. For other equity instruments, on the day of acquisition the Company can make an irrevocable election (on an instrument-by-instrument basis) to designate them as at FVTOCI. Financial liabilities are measured at amortized cost, unless they are required to be measured at FVTPL (such as instruments held for trading or derivatives) or if the Company has opted to measure them at FVTPL
Measurement
Financial assets and liabilities at amortized cost
Financial assets and liabilities at amortized cost are initially recognized at fair value plus or minus transaction costs, respectively, and subsequently carried at amortized cost less any impairment. The Company’s financial assets measured at amortized cost are comprised of its cash. The Company’s financial liabilities measured at amortized cost are comprised of its accounts payable and accrued liabilities.
Financial assets and liabilities at FVTPL
Financial assets and liabilities carried at FVTPL are initially recorded at fair value and transaction costs are expensed in the statements of loss and comprehensive loss. Realized and unrealized gains and losses arising from changes in the fair value of the financial assets and liabilities held at FVTPL are included in the statements of loss and comprehensive loss in the period in which they arise.
Debt instruments at FVTOCI
These assets are initially measured at fair value. Interest income calculated using the effective interest method, foreign exchange gains and losses and impairment are recognized in profit or loss. Other net gains and losses associated with changes in fair value are recognized in OCI. On derecognition, gains and losses accumulated in OCI are reclassified to profit or loss. The Company does not hold any debt instruments at FVTOCI.
Equity instruments at FVTOCI
These assets are initially measured at fair value. Dividends are recognized as income in profit or loss unless the dividend clearly represents a recovery of part of the cost of the investment. Other net gains and losses associated with changes in fair value are recognized in OCI and are never reclassified to profit or loss. The Company does not hold any equity instruments at FVTOCI.
Impairment of financial assets amortized at cost
The Company recognizes a loss allowance for expected credit losses on financial assets that are measured at amortized cost. At each reporting date, the Company measures the loss allowance for the financial asset at an amount equal to the lifetime expected credit losses if the credit risk on the financial asset has increased significantly since initial recognition. If at the reporting date, the financial asset has not increased significantly since initial recognition, the Company measures the loss allowance for the financial asset at an amount equal to the twelve month expected credit losses. The Company shall recognize in the statements of loss and comprehensive loss, as an impairment gain or loss, the amount of expected credit losses (or reversal) that is required to adjust the loss allowance at the reporting date to the amount that is required to be recognized.
F-14
Derecognition
Financial assets
The Company derecognizes financial assets only when the contractual rights to cash flows from the financial assets expire, or when it transfers the financial assets and substantially all of the associated risks and rewards of ownership to another entity.
Financial liabilities
The Company derecognizes a financial liability when its contractual obligations are discharged or cancelled or expire. The Company also derecognizes a financial liability when the terms of the liability are modified such that the terms and/or cash flows of the modified instrument are substantially different, in which case a new financial liability based on the modified terms is recognized at fair value.
Gains and losses on derecognition are generally recognized in profit or loss.
Note 3 — Critical estimates and judgments
The preparation of financial statements in accordance with IFRS requires the Company to use judgment in applying its accounting policies and make estimates and assumptions about reported amounts at the date of the financial statements and in the future. The Company’s management reviews these estimates and underlying assumptions on an ongoing basis, based on experience and other factors, including expectations of future events that are believed to be reasonable under the circumstances. Revisions to estimates are adjusted for prospectively in the period in which the estimates are revised.
Useful lives of property and equipment - Estimates of the useful lives of property and equipment are based on the period over which the assets are expected to be available for use. The estimated useful lives are reviewed annually and are updated if expectations differ from previous estimates due to physical wear and tear, technical or commercial obsolescence, not electing to exercise renewal options on Leases, and legal or other limits on the use of the relevant assets. In addition, the estimation of the useful lives of the relevant assets may be based on internal technical evaluation and experience with similar assets. It is possible, however, that future results of operations could be materially affected by changes in the estimates brought about by changes in the factors mentioned above. The amounts and timing of recorded expenses for any period would be affected by changes in these factors and circumstances. A reduction in the estimated useful lives of the property and equipment would increase the recorded expenses and decrease the non-current assets.
Estimating the incremental borrowing rate on leases - The Company cannot readily determine the interest rate implicit in leases where it is the lessee. As such, it uses its incremental borrowing rate (“IBR”) to measure lease liabilities. The IBR is the rate of interest that the Company would have to pay to borrow over a similar term, and with a similar security, the funds necessary to obtain an asset of comparable value to the right-of-use asset in a similar economic environment. IBR therefore reflects what the Company “would have to pay”, which requires estimation when no observable rates are available or where the applicable rates need to be adjusted to reflect the terms and conditions of the lease. The Company estimates the IBR using observable inputs (such as market interest rates) when available and is required to make certain entity-specific estimates.
Estimating the fair value of share-based payment transactions - The Company utilizes a Black-Scholes model to estimate the fair value of its share-based payments. In applying these models, management must estimate the expected future volatility of the Company’s estimated share price, and makes such assumptions based on a proxy of publicly-listed entities under an expectation that historical volatility is representative of the expected future volatility.
Other significant judgments - The preparation of these financial statements in accordance with IFRS requires the Company to make judgments, apart from those involving estimates, in applying accounting policies. The most significant judgments in applying the Company’s financial statements include:
F-15
Note 4 — Revenue from contracts with customers
Disaggregation of revenue from contracts with customers
The principal revenue activities of the Company for the years ended March 31, 2024, 2023 and 2022 were as follows:
|
|
For the Year Ended March 31, |
|
|||||||||
Revenue categories |
|
2024 |
|
|
2023 |
|
|
2022 |
|
|||
Revenue from ViraxClear |
|
|
|
|
|
|
|
|
— |
|
||
Total Revenue |
|
$ |
|
|
$ |
|
|
$ |
— |
|
||
Revenues were $
Accounting policies and significant judgments
Management does not consider there to be any significant judgments or estimates in the revenue recognition for the years ended March 31, 2024 and 2023.
Note 5 — Income tax
A reconciliation of income taxes at statutory rates with the reported taxes is as follows:
|
|
For the Year Ended March 31, |
|
|||||||||
|
|
2024 |
|
|
2023 |
|
|
2022 |
|
|||
Earnings (loss) for the year |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
Expected income tax (recovery) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Change in statutory, foreign tax, foreign exchange rates and other |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
Permanent Difference |
|
|
|
|
|
|
|
|
|
|||
Provision to NOLs |
|
|
|
|
|
— |
|
|
|
— |
|
|
Change in unrecognized deductible temporary differences |
|
|
|
|
|
|
|
|
|
|||
Total income tax expense (recovery) |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
As of March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Deferred Tax Assets (liabilities) |
|
|
|
|
|
|
||
Intangible assets |
|
|
|
|
|
— |
|
|
Depreciation |
|
|
( |
) |
|
|
— |
|
Non-capital losses available for future period |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Unrecognized deferred tax assets |
|
|
( |
) |
|
|
( |
) |
Net deferred tax asset (liability) |
|
$ |
— |
|
|
$ |
— |
|
The significant components of the Company’s temporary differences, unused tax credits and unused tax losses that have not been included on the consolidated statement of financial position are as follows:
|
|
March 31, 2024 |
|
|
Expiry Date Range |
|
March 31, 2023 |
|
|
Expiry Date Range |
||
Temporary Differences |
|
|
|
|
|
|
|
|
|
|
||
Non-capital losses available for future period - finite |
|
|
|
|
|
|
|
|
||||
Non-capital losses available for future period |
|
|
|
|
|
|
|
|
||||
Tax attributes are subject to review, and potential adjustment, by tax authorities.
F-16
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes.
Significant estimates — recognition of deferred tax assets
Deferred tax assets are recognized only to the extent that it is probable that the associated deductions will be available for use against future profits and that there will be sufficient future taxable profit available against which the temporary differences can be utilized, provided the asset can be reliably quantified. In estimating future taxable profit, management used “base case” approved forecasts which incorporate a number of assumptions, including a prudent level of future contracted revenue in the forecast period. In arriving at a judgment in relation to the recognition of deferred tax assets, management considers the regulations applicable to tax and advice on their interpretation. Future taxable income may be higher or lower than estimates made when determining whether it is appropriate to record a tax asset and the amount to be recorded. Furthermore, changes in the legislative framework or applicable tax case law may result in management reassessing the recognition of deferred tax assets in future periods.
At March 31, 2024 and 2023, there is an unrecognized deferred tax asset from net operating losses of $
The net operating losses in China can be carried forward up to
The net operating losses in Singapore may be carried forward indefinitely in general, subject to compliance with a shareholding test. Losses and unutilized capital allowances may be carried back for
Uncertain Tax Positions
The Company did not have significant unrecognized uncertain tax positions, or any unrecognized liabilities, interest or penalties associated with unrecognized tax benefit as of and for the years ended March 31, 2024 and 2023.
Note 6 — Loss per share
|
|
As of March 31, |
|
|||||||||
|
|
2024 |
|
|
2023 |
|
|
2022 |
|
|||
Loss for the year attributable to Virax |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Basic and diluted loss per share attributable to Virax – ordinary shares |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Basic loss per share is calculated by dividing the loss for the year by the weighted average number of ordinary shares in issue during the financial year. This calculation takes into effect the Share Consolidation as discussed in Note 18.
Diluted loss per share
Diluted loss per share is calculated by adjusting the weighted average number of ordinary shares in issue during the year to assume conversion of all dilutive potential ordinary shares. The Company had
|
|
As of March 31, |
|
|||||||||
|
|
2024 |
|
|
2023 |
|
|
2022 |
|
|||
Weighted average number of ordinary shares used in basic and diluted loss per share |
|
|
|
|
|
|
|
|
|
|||
Weighted average number of ordinary shares and potential ordinary shares used |
|
|
|
|
|
|
|
|
|
|||
F-17
Note 7 — VAT Receivable
|
|
For the Year Ended March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Unfiled VAT receivable |
|
$ |
|
|
$ |
— |
|
|
Filed VAT receivable |
|
|
|
|
|
— |
|
|
Total VAT receivable |
|
$ |
|
|
$ |
— |
|
|
Total VAT receivable as of March 31, 2024 consisted of $
Note 8 — Inventories
|
|
As of March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Finished goods |
|
|
|
|
|
|
||
Inventory write down |
|
|
— |
|
|
|
( |
) |
Inventories, net |
|
$ |
|
|
$ |
— |
|
|
Inventories as of March 31, 2024 consisted of ViraxClear and ViraxVet test kits stored in a warehouse operated by a third party. There was
Note 9 — Prepaid Expenses and Deposits
|
|
As of March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Prepaid directors and officers insurance |
|
|
|
|
|
|
||
Prepaid Nasdaq fee |
|
|
|
|
|
|
||
Prepaid vendor products |
|
|
|
|
|
|
||
Deposits |
|
|
|
|
|
|
||
Prepaid software subscription |
|
|
|
|
|
|
||
Other |
|
|
|
|
|
|
||
Prepaid expenses and deposits |
|
$ |
|
|
$ |
|
||
F-18
Note 10 — Property, plant & equipment, net
|
|
Lab Equipment |
|
|
Computer Equipment |
|
|
Capitalized Software |
|
|
Furniture & Fixtures |
|
|
Total |
|
|||||
Cost |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
Balance as of March 31, 2023 |
|
$ |
— |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
|||
Additions |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
Disposals |
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Effects of currency translation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Balance as of March 31, 2024 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
Accumulated Depreciation |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
Balance as of March 31, 2023 |
|
$ |
— |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
|||
Depreciation |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
Disposals |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
( |
) |
||
Effects of currency translation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Balance as of March 31, 2024 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|||
Depreciation expense for the year ended March 31, 2024 was $
Note 11 — Intangible Software, net
The Company capitalized certain costs associated with the ViraxImmune mobile application, which development has been suspended and has not been placed into services as of the date of this report. The Company capitalized $
The recoverable amount of the ViraxImmune mobile application has been calculated with reference to the following:
Based on the estimated projections, the Company concluded that since the ViraxImmune mobile application development has been suspended, and the present value of future cash flows does not exceed the carrying value of the capitalized amount, there is a full impairment of these amounts at March 31, 2024.
|
|
Intangible Software |
|
|
March 31, 2022 |
|
$ |
— |
|
Additions |
|
|
|
|
Impairment |
|
|
— |
|
March 31, 2023 |
|
|
|
|
Additions |
|
|
|
|
Impairment |
|
|
( |
) |
March 31,2024 |
|
$ |
— |
|
The following represents the details of the software development costs during the year:
Intangible Assets Under Development |
|
Type of Intangible Asset |
|
Net Book Value |
|
|
Remaining Amortization Period |
|
|
|
$ |
|
|
n/a |
|||
F-19
Note 12 — Accounts payable and accrued liabilities
|
|
As of March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Accounts payable |
|
|
|
|
|
|
||
Accrued bonuses |
|
|
— |
|
|
|
|
|
Accrued payroll taxes |
|
|
|
|
|
|
||
Accrued liabilities |
|
|
|
|
|
|
||
Total accounts payable and accrued liabilities |
|
$ |
|
|
$ |
|
||
Amounts included in accounts payables
Accounts payable for the year ended March 31, 2024 consists mostly of routine operating costs. Accrued payroll taxes for the year ended March 31, 2024 consisted of payroll taxes. Accrued liabilities for the year ended March 31, 2024 consists mainly of unpaid professional services. Accounts payable and accrued liabilities for the year ended March 31, 2023 consisted mainly of professional fees, legal fees and consulting services to various vendors.
Note 13 — Note Payable
On July 1, 2022, the Company entered into a note payable with a third party for the purpose of financing its Directors and Officers insurance policy. The unsecured loan at inception was $
Note 14 — Deferred Revenue
|
|
As of March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Deferred Revenue |
|
|
— |
|
|
|
|
|
Total Deferred Revenue |
|
$ |
— |
|
|
$ |
|
|
Deferred revenue is recorded when money is received in advance for our products that have not been fulfilled. For the year ended March 31, 2024, the Company recorded deferred revenue of $
Note 15 — Leases
Operating leases
Our leases consist of office and lab spaces with lease terms between
In May 2023, the Company entered into a lab space lease at BioCity for
The annual rent as of March 31, 2024 is as follows:
|
|
To June 30, 2024 |
GBP |
To June 30, 2025 |
GBP |
To June 30, 2026 |
GBP |
To June 30, 2027 |
GBP |
To June 30, 2028 |
GBP |
F-20
In addition to the annual rent, insurance is approximately GBP
In August 2023, the Company exited the old Shanghai lease which was a short term lease and not accounted for under IFRS 16 - Leases. We entered into a new office space lease in Shanghai for
Of these leases, our lab space at BioCity and office space in Shanghai are accounted for under IFRS 16 - Leases. We currently do not have leases with residual value guarantees or leases not yet commenced to which we are committed. Lease liabilities have been measured by discounting future lease payments using our incremental borrowing rate of
The following table summarizes our right-of-use assets activity for the BioCity and Shanghai lease the years ended March 31, 2024 and 2023, respectively.
|
|
Office and Lab Leases |
|
|
Cost |
|
|
|
|
Balance at March 31, 2022 |
|
$ |
— |
|
Additions |
|
|
— |
|
Effects of currency translation |
|
|
— |
|
Balance at March 31, 2023 |
|
$ |
|
|
Additions |
|
|
|
|
Effects of currency translation |
|
|
— |
|
Balance at March 31, 2024 |
|
$ |
|
|
|
|
|
|
|
Accumulated amortization |
|
|
|
|
Balance at March 31, 2022 |
|
$ |
— |
|
Amortization |
|
|
— |
|
Effects of currency translation |
|
|
— |
|
Balance at March 31, 2023 |
|
$ |
— |
|
Amortization |
|
|
( |
) |
Effects of currency translation |
|
|
— |
|
Balance at March 31, 2024 |
|
$ |
( |
) |
|
|
|
|
|
Right-of-use assets, net |
|
$ |
|
|
The following table summarizes the Company's lease liability activity for the BioCity lease and Shanghai lease for the years ended March 31, 2024 and 2023, respectively.
|
|
Total |
|
|
Balance as of March 31, 2022 |
|
$ |
— |
|
Additions |
|
|
— |
|
Payment of lease liabilities |
|
|
— |
|
Interest expense on lease liabilities |
|
|
— |
|
Effects of currency translation |
|
|
— |
|
Balance as of March 31, 2023 |
|
$ |
|
|
Additions |
|
|
|
|
Payment of lease liabilities |
|
|
( |
) |
Interest expense on lease liabilities |
|
|
|
|
Effects of currency translation |
|
|
— |
|
Balance as of March 31, 2024 |
|
$ |
|
|
The following table summarizes the maturity of our lease liabilities as of March 31, 2024:
F-21
|
|
March 31, 2024 |
|
|
Less than one year |
|
$ |
|
|
One to five years |
|
|
|
|
More than five years |
|
|
— |
|
Total lease payments |
|
$ |
|
|
Less: imputed interest |
|
|
( |
) |
Lease Liabilities |
|
$ |
|
|
Note 16 — Related party transactions
|
|
As of March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Related Party Payables |
|
|
|
|
|
|
||
James Foster |
|
|
— |
|
|
|
|
|
Cameron Lee Shaw |
|
|
— |
|
|
|
|
|
Total Related Party Payables |
|
$ |
— |
|
|
$ |
|
|
F-22
For the year ended March 31, 2022, the Company issued 17,253 ordinary shares for the settlement of related party payables amounting to $452,861.
For the year ended March 31, 2022 , the Company settled $7,906 of accrued interest to Fiona a Foster by issuance of
For the year ended March 31, 2024, there were
Related party compensation for the Company's Officers and Directors for the years ended March 31, 2024, 2023 and 2022 are as follows:
|
|
Year |
|
Salary ($) |
|
|
Bonus ($) |
|
|
Option Awards ($) (1) |
|
|
Other ($) (2) |
|
|
Total ($) |
|
|||||
James Foster, Chief Executive Officer and Director |
|
2024 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
2023 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
|
|
2022 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Cameron Shaw, Former Chief Operating Officer (3) |
|
2024 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|||
|
|
2023 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
|
|
2022 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|||
Nigel McCracken, Chief Operating Officer and Director (3) |
|
2024 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
2023 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
2022 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Dr. Tomasz George, Chief Scientific Officer (4) |
|
2024 |
|
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
||||
|
|
2023 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
|
|
2022 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Mark Ternouth, Chief Technical Officer and Director (5) |
|
2024 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
|
|
2023 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
|
|
2022 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Jason Davis, Chief Financial Officer |
|
2024 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
2023 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
|
|
2022 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Evan Norton, Independent Director |
|
2024 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
|
|
2023 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
|
|
2022 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Yair Erez, Independent Director |
|
2024 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
|
|
2023 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
|
|
2022 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Nelson Haight, Independent Director |
|
2024 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
|
|
2023 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
|
|
2022 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
(1) These amounts represent the aggregate grant fair value of stock options granted in the year ended March 31, 2024 and 2023, calculated in accordance with IFRS 2 “Share-based payment". Assumptions used in the calculation of these amounts are discussed in Note 18.
(2)These amounts represent employee benefits paid on behalf of the Company such as pension and health insurance. For Cameron Shaw, this amount consists of vacation and severance payout.
(3)Cameron Shaw departed the Company in September 2023 and is no longer an Officer or Director at March 31, 2024. All option award expense were reversed. Nigel McCraken, the current Chief Operating Officer, joined the Company on September 1, 2023.
(4)Dr. Tomasz George departed the Company in April 2024.
(5) Mark Ternouth departed the Company in June 2024.
F-23
Note 17 — Risk management overview
The Company has exposure to credit, liquidity and market risks from its use of financial instruments. This note provides information about the Company’s exposure to each of these risk, the Company’s objectives, policies and processes for measuring and managing risk. Further quantitative disclosures are included throughout these consolidated financial statements.
Credit risk
Credit risk is the risk of financial loss to the Company if a counterparty to a financial instrument fails to meet its contractual obligations and arises principally from the Company’s cash held with banks and other financial intermediaries.
The carrying amount of the cash represents the maximum credit exposure which amounted to $
Market risk
Market risk is the risk that changes in market conditions, such as commodity prices, foreign exchange rates, and interest rates, will affect the Company’s net income or the value of financial instruments. The objective of market risk management is to manage and control market risk exposures within acceptable limits, while maximizing the Company’s returns.
Foreign exchange risk
Foreign currency exchange rate risk is the risk that the fair value of future cash flows will fluctuate as a result of changes in foreign exchange rates. The Company does not currently use foreign exchange contracts to hedge its exposure to currency rate risk as management has determined that this risk is not significant at this point in time. As such, the Company’s financial position and financial results may be adversely affected by the unfavorable fluctuations in currency exchange rates.
As at March 31, 2024 and 2023, the Company had the following monetary assets and liabilities denominated in foreign currencies:
|
|
As of March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
|
|
RMB |
|
|
RMB |
|
||
Cash |
|
|
|
|
|
|
||
AP and Accrued Liabilities |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
||
|
|
As of March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
|
|
GBP |
|
|
GBP |
|
||
Cash |
|
|
|
|
|
|
||
AP and Accrued Liabilities |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
||
|
|
As of March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
|
|
EUR |
|
|
EUR |
|
||
Cash |
|
|
|
|
|
|
||
AP and Accrued Liabilities |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
||
|
|
As of March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
|
|
SGD |
|
|
SGD |
|
||
Cash |
|
|
|
|
|
|
||
AP and Accrued Liabilities |
|
|
( |
) |
|
|
( |
) |
Liquidity risk
The accompanying financial statements have been prepared on the basis of continuity of operations, realization of assets and the satisfaction of liabilities and commitments in the ordinary course of business.
F-24
Liquidity is the ability of a company to generate funds to support its current and future operations, satisfy its obligations, and otherwise operate on an ongoing basis. At March 31, 2024, the Company had a cash balance of $
Until such time that the Company implements its growth strategy, it expects to continue to generate operating losses in the foreseeable future, mostly due to research and development activities, corporate overhead and costs of being a public company.
Concentration risk
There was $
Capital Management
The Company aims to manage its capital resources to ensure financial strength and to maximize its financial flexibility by maintaining strong liquidity and by utilizing alternative sources of capital including equity, debt and bank loans or lines of credit to fund continued growth. The Company sets the amount of capital in proportion to risk and based on the availability of funding sources. The Company manages the capital structure and makes adjustments to it in light of changes in economic conditions and the risk characteristics of the underlying assets. As an early-stage growth company, the sale of ordinary shares has been the primary source of capital to date. Additional debt and/or equity financing may be pursued in future as deemed appropriate to balance debt and equity. To maintain or adjust the capital structure, the Company may issue new shares, take on additional debt or sell assets to reduce debt.
Note 18 — Stockholder’s equity
On June 19, 2022, the Company underwent a shareholding restructuring whereby the Company’s dual class share capital was amended to a single class of Ordinary Shares where
Share Consolidation
Beginning with the opening of trading on December 18, 2023, the Company's ordinary shares began trading on a post-Share Consolidation basis on the Nasdaq Capital Market under the same symbol "VRAX", but under a new CUSIP number of G9495L125. The objective of the Share Consolidation was to enable the Company to regain compliance with Nasdaq Marketplace Rule 5550(a)(2) and maintain its listing on the Nasdaq Capital Market. On January 4, 2024, the Company received notification that it had regained compliance with Nasdaq Marketplace Rule 5550(a)(2).
Authorized
As of March 31, 2024, there are a total of
Ordinary shares
Year ended March 31, 2022
For the year ended March 31, 2022, the Company issued
F-25
For the year ended March 31, 2022, the Company issued
For the year ended March 31, 2022, the Company entered into a Deed of Surrender with a shareholder relating to the balance of $
Year ended March 31, 2023
On May 10, 2022, we issued
On May 13, 2022, we issued
Initial Public Offering - On July 25, 2022, the Company consummated its IPO of
On August 5, 2022 Boustead Securities, LLC exercised
November Private Placement - On November 8, 2022, the Company entered into the November Securities Purchase Agreement ("SPA") with the Purchaser for the November Private Investment in Public Entity ("PIPE"), pursuant to which the Company received gross proceeds of approximately $
In December 2022, the Company issued
March Private Placement - On March 8, 2023, the Company entered into the March SPA with the same Purchaser for the March PIPE, pursuant to which the Company received gross proceeds of approximately $
Year ended March 31, 2024
In July 2023, pursuant to a settlement agreement between Boustead Securities LLC and the Company, the Company issued
On October 11, 2023, the “Company entered into an inducement offer letter agreement (the “Inducement Letter”) with a certain holder (the “Holder”) of
F-26
exercise price of $
As of March 31, 2024, the Company had
Warrants
The following summarizes activity related to the Company's outstanding warrants for the years ended March 31, 2024, 2023 and 2022:
|
Warrants |
|
|
Weighted Average Exercise Price |
|
|
Weighted Average Remaining Contractual Life (Years) |
|
|||
At March 31, 2021 |
|
— |
|
|
|
— |
|
|
|
— |
|
Granted |
|
— |
|
|
|
— |
|
|
|
— |
|
Exercised |
|
— |
|
|
|
— |
|
|
|
— |
|
Cancelled |
|
— |
|
|
|
— |
|
|
|
— |
|
At March 31, 2022 |
|
— |
|
|
|
— |
|
|
|
— |
|
Granted |
|
|
|
$ |
|
|
|
— |
|
||
Exercised |
|
( |
) |
|
$ |
— |
|
|
|
— |
|
Cancelled |
|
( |
) |
|
$ |
|
|
|
— |
|
|
At March 31, 2023 |
|
|
|
$ |
|
|
|
|
|||
Granted |
|
|
|
$ |
|
|
|
— |
|
||
Exercised |
|
( |
) |
|
$ |
|
|
|
— |
|
|
Cancelled |
|
— |
|
|
$ |
— |
|
|
|
— |
|
At March 31, 2024 |
|
|
|
$ |
|
|
|
|
|||
Warrants exercisable as of March 31, 2024 |
|
|
|
$ |
|
|
|
|
|||
Stock-based Compensation
The Company adopted the 2022 Equity Incentive Plan (the "2022 Plan") on March 15, 2022 and the 2023 Equity Incentive Plan (the "2023 Plan") on February 21, 2023, together, "the Plans". The Plans are intended to provide incentives which will attract and retain highly competent persons at all levels as employees of the Company, as well as independent contractors providing consulting or advisory services to the Company, by providing them opportunities to acquire the Company’s common stock or to receive monetary payments based on the value of such shares pursuant to awards issued. The 2022 Plan permits the grant of options and shares for up to
The Company uses the Black-Scholes option-pricing model to estimate the fair value of its stock option awards. The calculation of the fair value of the awards using the Black-Scholes option-pricing model is affected by the Company’s stock price on the date of grant as well as assumptions regarding the following:
|
As of March 31, 2024 |
|
As of March 31, 2023 |
|
As of March 31, 2022 |
|
|||
Expected volatility |
|
|
|
— |
|
||||
Expected term |
|
|
|
— |
|
||||
Risk-free interest rate |
|
|
|
— |
|
||||
Dividend yield |
|
— |
|
|
— |
|
|
— |
|
Forfeiture rate |
|
|
|
— |
|
||||
Note there were
The expected volatility was determined with reference to the historical volatility of the Company’s stock. The Company uses historical data to estimate option exercise and employee termination within the valuation model. The expected term of options granted represents the period of time that options granted are expected to be outstanding. The risk-free interest rate for periods within the contractual life of the option is based on the U.S. Treasury rate in effect at the time of grant.
F-27
In April 2023, an aggregate of
As the modified grants were all unvested at the time of the modification, the incremental charge is accrued in the modification period based on the amortization of outstanding shares as of the modification date, with the remaining incremental expense recognized over the period of the remaining vesting period, in line with the remaining amortization of the original fair value.
For the year ended March 31, 2024, the Company issued stock options to purchase
For the years ended March 31, 2024 and 2023, the Company recognized an expense of $
A summary of the status of the Company’s outstanding stock options as of March 31, 2024 and changes during the periods ending on that date is as follows:
|
Shares |
|
|
Exercise Price |
|
|
Grant Date Fair Value |
|
|
Aggregate Intrinsic Value |
|
|
Weighted Average Remaining Term (Years) |
|
|||||
Options |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
At March 31, 2021 |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Granted |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Exercised |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Expired |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Forfeiture |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
At March 31, 2022 |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Granted |
|
|
|
$ |
|
|
$ |
|
|
|
— |
|
|
|
|
||||
Exercised |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Expired |
|
( |
) |
|
$ |
|
|
$ |
|
|
|
— |
|
|
|
— |
|
||
Forfeiture |
|
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
— |
|
|
|
— |
|
At March 31, 2023 |
|
|
|
$ |
|
|
$ |
|
|
|
— |
|
|
|
|
||||
Granted |
|
|
|
$ |
|
|
$ |
|
|
|
— |
|
|
|
|
||||
Exercised |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Expired |
|
( |
) |
|
$ |
|
|
$ |
|
|
|
— |
|
|
|
— |
|
||
Forfeiture |
|
( |
) |
|
$ |
|
|
$ |
|
|
|
— |
|
|
|
— |
|
||
At March 31, 2024 |
|
|
|
$ |
|
|
$ |
|
|
|
— |
|
|
|
|
||||
Exercisable at March 31, 2024 |
|
|
|
$ |
|
|
$ |
|
|
|
— |
|
|
|
|
||||
Note 19 — Subsequent Events
On April 18, 2024 the Company granted
F-28
On June 7, 2024, the Company sold
F-29